

General5 min read
신약 발견부터 상업 운영까지: AmpleLogic이 전체 제약 가치 사슬을 강화하는 방법
대부분의 제약 회사는 연구, 품질, 제조 및 규제 기능 전반에 걸쳐 5~10개의 분리된 시스템을 실행합니다. 단일 연결된 플랫폼이 전체 제품 라이프사이클에 걸쳐 가능한 변화를 어떻게 변화시키는지는 다음과 같습니다.
2026-07-30더 읽기
제약 제조, 컴플라이언스, AI 자동화 및 생명과학 디지털 전환에 관한 전문가 관점.
대부분의 제약 회사는 연구, 품질, 제조 및 규제 기능 전반에 걸쳐 5~10개의 분리된 시스템을 실행합니다. 단일 연결된 플랫폼이 전체 제품 라이프사이클에 걸쳐 가능한 변화를 어떻게 변화시키는지는 다음과 같습니다.

인도 약전 제10판에는 121개의 새로운 논문이 추가되었으며 기존 실험실 시스템에서는 추적할 수 없었던 롤링 개정 일정이 추가되었습니다. IP 2026이 QC 실험실에 미치는 영향과 이에 발맞추기 위해 디지털 품질 시스템이 필수적인 이유는 다음과 같습니다.

수동 액세스 제어는 LIMS, eQMS, eBMR 및 기타 검증된 시스템 전체에서 확장되지 않습니다. 검사관이 규정 준수 격차를 발견하기 전에 이를 해소하기 위해 GAMP 호환 역할 기반 사용자 액세스 관리 시스템이 필수적인 이유는 다음과 같습니다.

전자 실험실 노트와 실험실 정보 관리 시스템은 서로 다른 목적으로 사용되지만 함께 연결된 실험실 환경을 만듭니다. ELN 소프트웨어와 LIMS 소프트웨어를 통합하여 효율성을 향상하고 데이터 무결성을 강화하며 제약 및 생명 과학 실험실의 규정 준수를 지원하는 방법을 알아보세요.

단편화된 스프레드시트와 레거시 도구는 글로벌 제약 규제 요구 사항을 따라잡을 수 없습니다. 제출, 감시, 규정 준수 추적을 포함하는 연결된 규제 정보 관리 시스템이 있으면 좋은 인프라가 아니라 필수 인프라가 된 이유는 다음과 같습니다.

최신 공정 검증 소프트웨어는 제약 제조업체가 수동 검증을 자동화된 규정 준수 워크플로로 대체하는 데 도움이 됩니다. AmpleLogic CVS가 추적성을 개선하고 승인을 가속화하며 검사 준비를 보장하는 동시에 검증 위험을 줄이는 방법을 알아보세요. GMP 준수 제조에 디지털 프로세스 검증이 필수적인 이유를 알아보세요.

제약 및 생명공학 연구소는 매년 더 빠르게 움직이도록 압력을 받고 있으며, 이러한 압력 중 어느 것도 규정 준수를 완화할 수 없습니다. 많은 팀이 여전히 종이 노트나 10년 동안 함께 패치된 레거시 소프트웨어에 의존하고 있다면 이는 어려운 일입니다. 차세대 전자 실험 노트(Electronic Laboratory Notebook)는 여기서 계산을 바꿉니다. 단순한 디지털 연구실 노트북이 아니라 과학 조직의 연결 계층으로 작동하기 시작하여 연구, 품질, 제조 및 규제 작업을 서로 소통하지 않는 일련의 별도 단계 대신 하나의 라이프사이클로 묶습니다.

일관된 제품 품질을 유지하는 것은 지속적인 프로세스 가시성에서 시작됩니다. AmpleLogic의 제약 프로세스 모니터링 소프트웨어가 제조업체가 CPV(지속적 프로세스 검증)를 통해 중요한 프로세스를 실시간으로 모니터링하고, 변동성을 줄이고, 규정 준수를 강화하고, 운영 효율성을 향상시키는 데 어떻게 도움이 되는지 알아보세요.

제약 공장은 여전히 종이 기반 추적, 산재된 기록, 대응적 유지 관리로 인해 시간과 감사 신뢰성을 잃고 있습니다. 교정 누락, 시스템 연결 끊김, 장비 상태에 대한 실시간 가시성 부족 등으로 인해 배치가 실패하거나 감사자가 질문을 시작한 후에만 문제가 표면화됩니다. AmpleLogic의 CAPS는 교정 및 유지 관리 일정을 자동화하고 GMP 준수, 21 CFR Part 11 정렬 감사 추적을 유지하고 eQMS, LIMS, eBMR/MES, DMS, LMS 및 SAP/ERP와 통합하여 검증된 장비만 생산에 도달하도록 하여 이 문제를 해결합니다. 그 결과, 긴급 수리 횟수가 줄어들고, 가동 중지 시간이 줄어들며, 혼란스러운 대신 보고서를 가져오는 감사 준비가 가능해집니다.

수동 스프레드시트는 글로벌 제약 규정을 따라갈 수 없습니다. 최신 규제 정보 관리 시스템이 제출, 추적 및 규정 준수 감시를 하나의 시스템으로 중앙 집중화하는 방법은 다음과 같습니다.

효율적인 실험실 운영은 효과적인 계획 수립에서 시작됩니다. 품질 관리 계획 소프트웨어가 제약 회사에서 QC 일정을 간소화하고 실험실 리소스를 최적화하며 AmpleLogic의 디지털 계획 솔루션을 통해 검사 준비 상태를 향상시키는 데 어떻게 도움이 되는지 알아보세요.

문서화는 의약품 제조의 근간입니다. 모든 배치, 모든 단계, 모든 편차 등 모든 항목은 검사 대상 국가에 관계없이 규제 기관이 만족할 수 있는 방식으로 기록, 확인 및 보관되어야 합니다. 수년 동안 이는 작업 현장에서 QA까지 이동한 다음 다시 돌아오고, 아마도 다시 한 번 다시 돌아오는 종이 배치 기록 더미를 의미했습니다. 효과가 있었습니다. 간신히. 그리고 어떤 시점에서는 균열이 너무 넓어져서 계속 패치를 할 수 없게 되었습니다. 이것이 바로 eBMR 소프트웨어가 등장하는 곳입니다. 선택적 업그레이드에서 공장 없이는 할 수 없는 것으로 조용히 사라진 전자 배치 제조 기록 시스템입니다.

거의 모든 제약 연구실에 가보면 10만 달러에 달하는 새로운 기기 바로 옆에 종이 일지가 놓여 있는 것을 발견할 가능성이 있습니다. 이상하게도 생각해보면. 연구실에서는 자동화와 분석에 돈을 쏟아 붓지만, 누가 언제 무엇을 했는지 기록하는 기본 일지는 여전히 펜이 끈으로 묶인 나선형 노트일 때가 많습니다.

품질 팀이 감사 전에 SOP의 "최종 최종 버전"을 찾기 위해 애쓰는 모습을 본 적이 있다면 문서 관리로 인해 규정 준수 리더가 밤을 새워야 하는 이유를 이미 알고 계실 것입니다. AmpleLogic의 문서 관리 시스템 소프트웨어는 모든 버전, 승인 및 서명이 검사 대상으로 유지되어야 하는 규제된 생명 과학 환경을 위해 특별히 제작되었습니다. 흩어져 있는 폴더와 수동 라우팅을 GMP 문서가 실제로 이동하는 방식에 맞게 설계된 GAMP 호환 감사 준비 플랫폼으로 대체합니다.

QA 전문가에게 매년 가장 두려운 것이 무엇인지 물어보면 "APQR 시즌"이 빠르게 다가옵니다. 거의 매번. 서류상으로는 연례 제품 품질 검토가 복잡해 보이지 않습니다. 1년간의 제조, 실험실 및 품질 데이터를 수집하세요. 분석해보세요. 귀하의 제품이 여전히 예상대로 작동하고 있음을 보여주십시오. 아주 간단하죠?

세척 공정 검증 소프트웨어가 제약 제조업체가 검증을 간소화하고 규정 준수 위험을 줄이며 운영 효율성을 향상시키는 데 어떻게 도움이 되는지 알아보세요. 현대 제조에서 데이터 무결성, 감사 준비 및 규정 준수를 유지하는 데 디지털 프로세스 검증 시스템을 채택하는 것이 필수적인 이유를 알아보세요.

거의 모든 제약 공장의 엔지니어링 또는 QA 담당자에게 교정을 요청하고 어떤 일이 일어나는지 지켜보세요. 일시 정지가 있습니다. 아마도 한숨일 것이다. 그러면 어떤 대답이 나오든 대개 "글쎄, 기술적으로는..."으로 시작합니다. 모두가 이 문제가 중요하다는 것을 알고 있으며, 누구도 그것에 대해 논쟁하지 않습니다. 하지만 대부분의 시설에서 실제로 이를 어떻게 처리하는지 확인하면 몇 년 동안 사용되어 온 것과 동일한 설정을 찾을 수 있습니다. 누군가가 2014년에 만든 스프레드시트, 바인더에 채워진 종이 로그, 팀 절반이 무시하는 달력 알림 등입니다.

지속적인 공정 검증은 제약 제조업체가 초기 검증 이후 공정 제어를 유지하는 데 도움이 됩니다. CPV 소프트웨어가 어떻게 지속적인 모니터링, 통계 분석, 프로세스 추세 조기 식별을 지원하는지 알아보세요.

일괄 릴리스는 예전에는 그렇게 큰 일이 아니었습니다. 대부분 서류 양식을 직접 작성하고 부서에서 부서로 전달하고 각 정류장에서 승인을 받은 다음 모든 것이 올바르게 확인되면 일괄 배송을 의미했습니다. 공장의 생산량이 적고 검사 빈도가 낮았던 시절에는 괜찮았습니다. 요즘은 그냥 참지 못합니다. 검사관들은 조립하는 데 일주일이 걸리는 바인더가 아니라 현장에서 검토할 수 있는 기록을 기대하며 나타납니다. 환자들은 이미 승인된 의약품을 예상보다 오랫동안 기다리며 갇혀 있습니다. 그리고 현재 많은 제조업체들이 여러 사이트를 동시에 운영하고 있는데, 이는 기본적으로 종이 양식과 희망으로는 추적이 불가능합니다. 따라서 바이오제약 업계 리더들 사이의 실제 대화는 더 이상 "일괄 출시를 현대화해야 하는가"가 아닙니다. 이는 규정 준수 측면에서 실수로 무언가를 위반하지 않고 중요할 만큼 빠르게 수행하는 것입니다.

최신 문서 제어 소프트웨어가 제약 회사가 버전 제어 문제, 승인 지연 및 감사 준비 문제를 극복하는 데 어떻게 도움이 되는지 알아보세요. 중앙 집중식 문서 관리 시스템이 규제 문서 워크플로에 더 큰 제어, 추적성 및 효율성을 제공하는 방법을 알아보세요.

오늘날 거의 모든 제약 연구실에 가보면 여전히 종이 노트, 인쇄된 프로토콜, 계기판에 붙어 있는 스티커 메모 더미를 발견할 수 있습니다. 정확하고 빈틈없는 데이터를 기반으로 운영되어야 하는 업계에서 보는 것은 이상한 일입니다. 과학자들은 결과를 손으로 기록한 다음 나중에 다른 시스템에 동일한 숫자를 다시 입력합니다. 그 과정 중 어딘가에서 실수가 발생합니다. 페이지가 누락됩니다. 그리고 감사관이 3년 전의 실험 기록을 요청하면 누군가는 오후 내내 보관 상자를 뒤지느라 시간을 보내게 됩니다.

대부분의 연구실에는 이미 LIMS가 있습니다. 실제 문제는 이것이 여전히 유지될 수 있는지 여부입니다. AI가 QC 실험실에서 안정성 예측, 재고 및 STP 처리를 어떻게 변화시키고 있는지 살펴보겠습니다.

품질 관리는 반응형에서 예측형으로 전환되고 있습니다. AI가 eQMS를 어떻게 변화시키고 있는지, 그리고 미래를 위해 구축된 플랫폼을 선택할 때 우선순위를 정해야 할 사항은 다음과 같습니다.

수동 QC 계획은 종종 일정 충돌, 배치 출시 지연, 실험실 효율성 감소로 이어집니다. 최신 **QC 관리 시스템**이 제약 실험실에서 계획을 간소화하고, 리소스를 최적화하고, 지능형 QC 일정을 통해 규정 준수를 유지하는 데 어떻게 도움이 되는지 알아보세요.

APQR 시즌 동안 대부분의 제약 품질 부서를 방문하면 동일한 일이 진행되는 것을 볼 수 있습니다. 누군가 세 개의 스프레드시트를 열어 놓고 근처에 배치 기록 더미가 있고 보고서 템플릿이 손으로 채워지기를 기다리고 있습니다. 연간 제품 품질 검토는 구조화된 데이터 기반 프로세스로 이루어져야 합니다. 실제로 많은 팀의 경우 여전히 매년 몇 주가 소요되는 수동 작업입니다. 데이터 양이 증가하고 규제 당국이 더 많은 것을 기대함에 따라 이러한 기존 작업 방식은 더 이상 유지되지 않습니다.

Walk into any pharma plant and you'll find the same quiet truth: everything depends on equipment doing exactly what it's supposed to do, every time, no exceptions. A scale that's drifted a little out of range. A sensor nobody got around to checking on schedule. A maintenance log stuffed in a drawer somewhere, half-filled and out of date. Any one of these can put a whole batch on the line. That's the thing about calibration and preventive maintenance, they look like engineering chores on the surface, but underneath, they're really compliance work. Miss one, and you're not just risking a machine breakdown, you're risking a regulatory finding.

최신 문서 관리 시스템(DMS)을 통해 제약 회사가 문서 제어를 간소화하고 규정 준수를 유지하며 검사 준비 상태를 유지하는 데 어떻게 도움이 되는지 알아보세요. 규정을 준수하는 제약 문서 관리 솔루션에서 찾아야 할 주요 기능을 알아보세요.

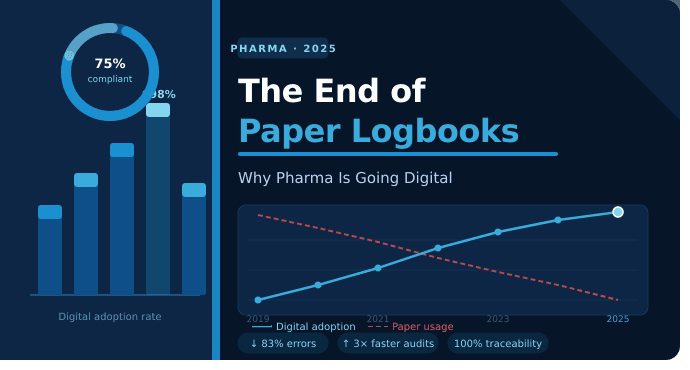
의약품 제조는 일부 지역에서는 빠르게 진행되고 다른 지역에서는 정체 상태를 유지하고 있습니다. 배치 기록은 디지털입니다. 실험실 장비는 LIMS와 통신합니다. 그러나 많은 작업 현장에서는 여전히 클립보드가 탱크나 HVAC 패널 옆에 매달려 있어 누군가가 직접 판독값을 기록하기를 기다리고 있습니다. 하이테크 시스템과 로우테크 로그북 사이의 격차는 대부분의 감사 문제가 시작되는 지점이며, AmpleLogic의 전자 로그북(eLogbook) 소프트웨어가 차이를 만들려고 노력하는 지점이기도 합니다.

오늘날 대부분의 제약 시설에 가보면 서류 캐비닛에 여전히 종이 배치 기록이 들어 있는 것을 볼 수 있습니다. 서명하고, 스탬프를 찍고, 검토하고, 저장했습니다. 그리고 그 사슬 어딘가에서 뭔가 잘못되었습니다. 누락된 서명입니다. 잘못된 항목입니다. 감사 전에 사라지는 페이지입니다. 이것은 예외가 아닙니다. 시설 관리보다 더 많은 일이 발생합니다.

최신 문서 관리 시스템 소프트웨어가 제약 회사의 규정 준수 개선, 문서 제어 간소화, 감사 준비 유지에 어떻게 도움이 되는지 알아보세요. 생명과학 산업에서 디지털 문서 관리가 품질, 효율성 및 규제 성공에 필수적인 이유를 알아보세요.

AI는 eQMS 플랫폼을 사후 파일링 시스템에서 사전 품질 파트너로 전환하고 있습니다. 무엇이 바뀌고, 무엇을 먼저 자동화해야 하는지, 그리고 실제 AI 기반 플랫폼과 유행어를 구분하는 요소는 다음과 같습니다.

MACO 계산, 위험 평가 및 검증 워크플로우를 자동화하는 AI 기반 세척 검증 소프트웨어로 의약품 규정 준수를 간소화하세요. 중앙 집중식 디지털 검증 플랫폼을 통해 감사 준비를 개선하고 수동 작업을 줄이며 규정 준수를 보장합니다.

연간 제품 품질 검토는 규제 요건이지만 대부분의 제약 품질팀에서는 조용히 가장 지치고 반복되는 작업 중 하나가 되었습니다. 프로세스는 본질적으로 수동입니다. 데이터는 단절된 시스템에 분산되어 있습니다. 배치 기록은 한 곳에, 불만사항은 다른 곳에, 안정성 데이터는 다른 곳에 있습니다. 누군가는 모든 제품에 대해 매년 반복적으로 모든 것을 종합하고, 조정하고, 형식을 지정하고, 일관성 있는 보고서로 바꿔야 합니다. 서류상으로는 규정 준수 연습입니다. 실제로는 수백 시간의 직원 시간이 다른 곳으로 갈 수 있습니다.

Pharma의 오래된 플레이북 스프레드시트, 고립된 데이터, 수동 분석은 최신 데이터 볼륨의 무게로 인해 무너지고 있습니다. 이번 에디션에서는 AI가 신약 발견, 임상 시험, 운영 및 시장 전략 전반에 걸쳐 인텔리전스를 어떻게 재배치하는지, 그리고 왜 다음 승리를 거두는 기업이 가장 많은 데이터를 보유한 기업이 아니라 이를 가장 빠르게 결정으로 전환하는 기업인지 살펴봅니다.

지속적인 공정 검증(CPV)은 제약 제조업체가 공정 제어를 유지하고 규정 준수를 보장하며 품질 위험이 생산에 영향을 미치기 전에 감지하는 데 도움이 됩니다. 최신 CPV 소프트웨어가 어떻게 실시간 모니터링, 자동화된 SPC 차트 작성, 지능형 경고 및 사전 품질 관리를 위한 APQR 자동화를 지원하는지 알아보세요.

제약 분야의 수동 안정성 연구 관리는 가져오기 날짜 누락, 데이터 격차 및 규정 준수 위험으로 이어집니다. 최신 안정성 테스트 소프트웨어는 일정 관리를 자동화하고 추적성을 개선하며 규제에 대비한 안정성 프로그램을 보장합니다.

지능형 QC 계획 소프트웨어가 제약 및 생명공학 실험실에서 일정 관리를 자동화하고, 리소스를 최적화하고, 규정 준수를 개선하고, 처리 시간을 단축하는 데 어떻게 도움이 되는지 알아보세요. 보다 스마트한 계획과 캠페인 기반 실행이 현대 QC 운영을 어떻게 변화시켜 효율성을 높이고 배치 릴리스를 더욱 빠르게 하는지 알아보세요.

모든 제약 감사에는 스토리가 있습니다. FDA 483 관찰 또는 MHRA 조사 도중에 조사관이 믿을 수 없을 정도로 단순해 보이는 질문을 하는 순간이 있습니다. "누가 이 시스템에 접근했는지, 언제였습니까?" 너무 많은 생명 과학 조직의 경우 해당 질문에 답변하는 데 며칠이 걸리거나 전혀 명확하게 답변할 수 없습니다. 자세한 내용과 특수 목적으로 구축된 GxP 액세스 관리의 실제 모습은 다음과 같습니다.

정시에 소프트웨어를 배송하는 것은 매우 중요합니다. 조직이 진정으로 차별화할 수 있는 정의된 품질 표준에 따라 구축되고 안정적이고 규정을 준수하는 소프트웨어를 제공합니다. 소프트웨어 품질 관리 시스템(SQMS)은 품질 계획 및 보증부터 지속적인 개선 및 규제 감사 준비에 이르기까지 전체 개발 라이프사이클에서 이를 실현할 수 있는 구조화된 프레임워크를 제공합니다.

수동 데이터 입력은 현대 실험실의 가장 큰 과제 중 하나로 남아 있으며, 이는 수기 오류, 규정 준수 위험 및 운영 비효율성을 초래합니다. 이 기사에서는 LIMS의 기기 통합이 어떻게 데이터 캡처를 자동화하고, 정확성을 개선하고, 규정 준수를 지원하고, 제약, 생명 과학, 식품 테스트 실험실 전반에서 생산성을 향상하는지 살펴봅니다.

제약, 생명 공학, 의료 기기 산업에서 문서를 관리하려면 엄격한 규정 준수, 보안 및 효율성이 필요합니다. AmpleLogic 문서 관리 시스템이 어떻게 문서 제어를 간소화하고, 워크플로를 자동화하며, 안전한 디지털 플랫폼을 통해 규정 준수를 보장하는지 알아보세요.


안정성 연구는 의약품이 수명주기 전반에 걸쳐 안전하고 효과적이며 규정을 준수하도록 보장하는 데 필수적입니다. 그러나 스프레드시트와 수동 프로세스를 통해 이러한 연구를 관리하면 규정 준수 위험, 데이터 무결성 문제 및 운영 비효율성이 발생할 수 있습니다. 이 블로그에서는 현대 디지털 안정성 프로그램의 주요 구성 요소인 엔드투엔드 안정성 관리의 중요성, 규제 기대치, 그리고 제약 조직이 AmpleLogic의 안정성 관리 플랫폼과 같은 중앙 집중식 규정 준수 솔루션을 사용하여 안정성 연구를 간소화할 수 있는 방법을 살펴봅니다.

AI 기반 전자 로그북은 디지털 기록 보관과 지능형 자동화를 결합하여 제약 문서를 혁신하고 있습니다. 데이터 무결성 및 규정 준수 개선부터 예측 통찰력 및 실시간 이상 감지 지원에 이르기까지 AI 기반 eLogbook은 제약 회사가 운영을 간소화하고 수동 작업을 줄이며 Pharma 4.0 이니셔티브를 가속화하는 데 도움이 됩니다.
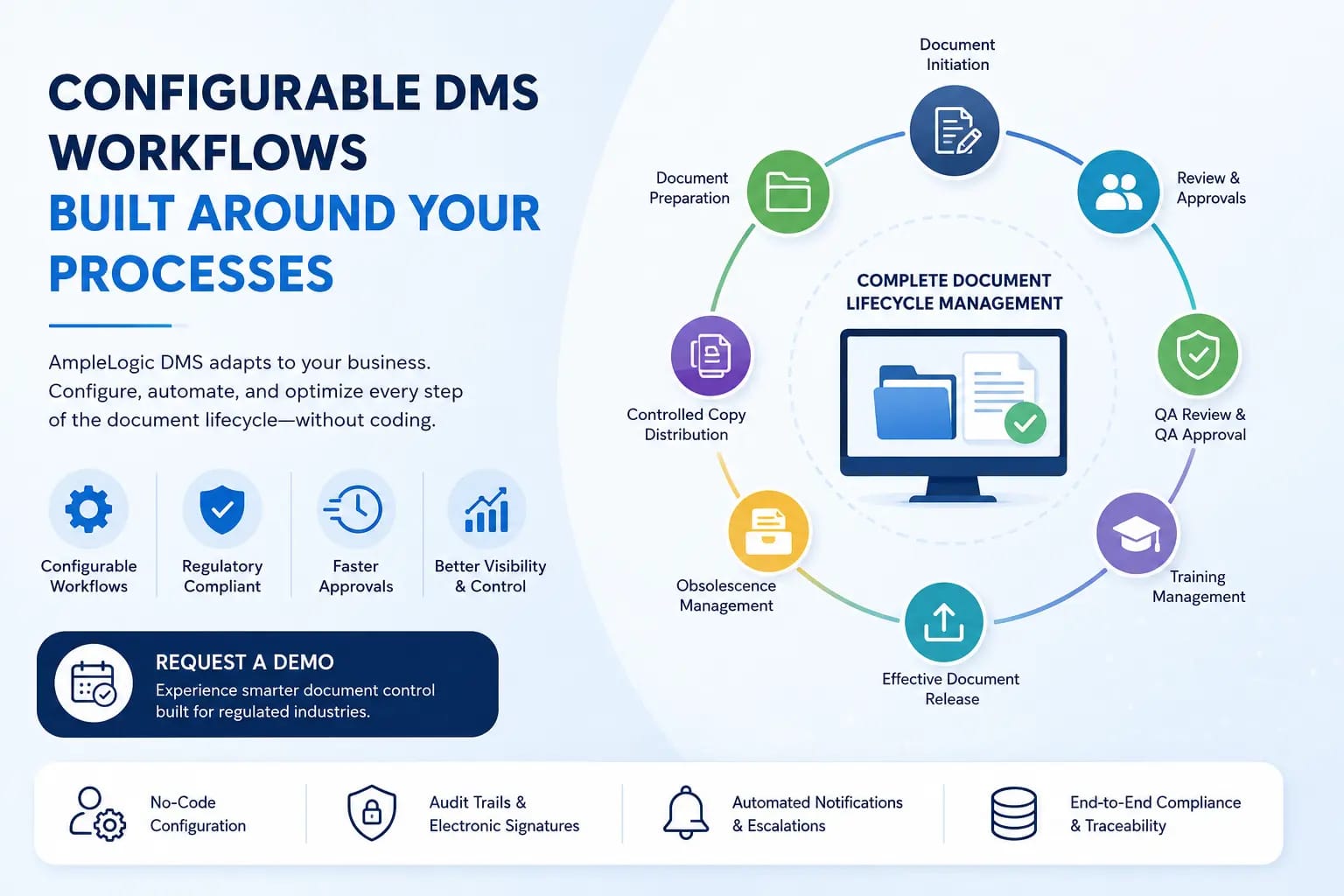
문서 작성 및 검토부터 교육, 정기 검토, 노후화 관리에 이르기까지 효율적인 워크플로우 자동화는 규정 준수 및 생산성을 위해 필수적입니다. AmpleLogic DMS가 규제 대상 산업을 위해 설계된 완전히 사용자 정의 가능한 워크플로우를 통해 승인 병목 현상을 제거하는 방법을 알아보세요.

품질은 더 이상 단순한 부서가 아니라 전략입니다. 제약, 제조, 생명 과학 전반에 걸쳐 규제 기대치가 강화됨에 따라 지능형 품질 관리 시스템 소프트웨어에 투자하는 조직은 단순히 규정을 준수하는 데 그치지 않습니다. 그들은 더 빠르고, 더 스마트하며, 훨씬 더 큰 자신감을 갖고 운영합니다. 이 가이드에서는 최신 QMS 플랫폼의 기능, 좋은 구현과 훌륭한 구현을 구분하는 요소, 업계 및 규모에 적합한 솔루션을 선택하는 방법을 자세히 설명합니다.

DMS 소프트웨어의 일반적인 버전 제어 문제에 대해 알아보고 문서 정확성, 규정 준수, 협업 및 감사 준비를 보장하는 효과적인 솔루션을 찾아보세요.

제약 공장의 예방 유지보수와 예측 유지보수의 차이점과 교정 및 유지보수 소프트웨어가 규정 준수, 장비 신뢰성 및 운영 효율성을 향상시키는 방법에 대해 알아보세요.

프로세스 표준화 및 시스템 통합부터 규정 준수 및 사용자 채택에 이르기까지 eBMR 구현 중에 제약회사가 직면하는 가장 큰 과제를 살펴보세요. 최신 eBMR 소프트웨어가 배치 제조를 간소화하고, 데이터 무결성을 개선하고, 제약 운영의 디지털 혁신을 가속화하는 데 어떻게 도움이 되는지 알아보세요.

종이 로그북에서 전자 시스템으로 전환하면 GMP 시설이 규정 준수를 개선하고 문서 오류를 줄이며 운영 효율성을 높이는 데 도움이 됩니다. 전자 일지가 어떻게 현대 제약 제조에서 데이터 무결성, 더 빠른 감사, 향상된 배치 출시 프로세스 및 측정 가능한 ROI를 지원하는지 알아보세요.

최신 LIMS 솔루션은 자동화, 실시간 가시성 및 향상된 데이터 무결성을 통해 제약 실험실이 OOS 및 OOT 조사를 간소화하는 데 도움이 됩니다. 디지털 조사 워크플로우가 규정 준수를 강화하고 근본 원인 분석을 가속화하며 전반적인 품질 관리를 강화하는 방법을 알아보세요.

AI Copilots가 품질 운영 전반에 걸쳐 더 스마트한 규정 준수, 더 빠른 조사, 예측 통찰력 및 지능형 자동화를 지원하여 제약 품질 관리를 어떻게 변화시키고 있는지 알아보세요. AI 기반 제약 소프트웨어가 조직의 효율성을 향상시키고 수작업을 줄이며 미래에 대비한 디지털 품질 시스템을 구축하는 데 어떻게 도움이 되는지 알아보세요.

AI 기반 세척 검증 소프트웨어가 제약 제조업체가 MACO 계산을 자동화하고, GMP 규정 준수를 개선하고, 수동 검증 비효율성을 제거하는 데 어떻게 도움이 되는지 알아보세요. 디지털 검증 워크플로우가 제약 제조 전반에 걸쳐 데이터 무결성, 감사 준비성 및 운영 효율성을 향상시키는 방법을 알아보세요.

기술과 비즈니스의 미래를 형성하는 획기적인 아이디어, 혁신 전략, 혁신적인 솔루션을 평가한 AL Ideathon 2024의 비전 있는 심사위원단을 만나보세요.



aPaaS(Application Platform as a Service)는 기본 인프라를 처리하지 않고도 애플리케이션을 구축, 배포 및 관리하기 위한 완전한 환경을 제공하는 클라우드 컴퓨팅 모델입니다. 이를 통해 기업은 개발을 가속화하고 비용을 절감하며 애플리케이션을 효율적으로 확장할 수 있습니다. 도구, 프레임워크 및 자동화를 제공함으로써 aPaaS는 설계부터 배포까지 전체 애플리케이션 수명주기를 단순화하여 조직이 디지털 우선 세계에서 더 빠르게 혁신하고 경쟁력을 유지할 수 있도록 돕습니다.

GAMP 5는 제약 산업의 컴퓨터 시스템을 검증하기 위한 구조화된 접근 방식을 제공하여 해당 시스템이 의도된 용도에 적합하고 규제 기대치에 부합하는지 확인합니다. 요구 사항부터 테스트까지 수명 주기 기반 검증 프로세스를 지원하는 V-Model과 같은 프레임워크와 함께 작동합니다. 21 CFR Part 11 및 EU Annex 11과 같은 규정은 전자 기록 및 서명에 대한 엄격한 규칙을 정의하여 시스템 전반에 걸쳐 데이터 무결성, 보안 및 추적성을 보장합니다. 이러한 표준은 조직이 검증된 소프트웨어 시스템을 효율적으로 개발, 구현 및 유지할 수 있는 강력한 규정 준수 생태계를 만듭니다. 위험 기반 검증, 수명주기 관리, 규제 조정을 결합함으로써 기업은 규정 준수 위험을 줄이고 제품 품질을 개선하며 제약 운영의 디지털 혁신을 가속화할 수 있습니다.

로우 코드 플랫폼을 사용하면 조직은 시각적 인터페이스와 사전 구축된 구성 요소를 사용하여 최소한의 수동 코딩으로 애플리케이션을 설계, 개발 및 배포할 수 있습니다. 제약 회사의 경우 규제 표준과 데이터 무결성을 유지하면서 프로세스를 디지털화하고, 워크플로를 자동화하고, 운영을 확장할 수 있는 더 빠르고 규정을 준수하는 방법을 제공합니다.

BMR(배치 기록) 발행 및 배치 번호 생성 소프트웨어는 제약 회사가 중요한 제조 프로세스를 자동화하고 제어하는 데 도움이 됩니다. 안전하고 추적 가능하며 효율적인 디지털 워크플로우를 제공하여 정확한 배치 추적을 보장하고 수동 오류를 줄이며 규제 표준 준수를 유지합니다.

제약 QA/QC 배치 프로세스 자동화를 통해 기업은 USFDA, MHRA 및 cGMP 표준을 준수하면서 품질 보증 및 제어 활동을 간소화할 수 있습니다. 워크플로를 디지털화하고 중요한 프로세스를 자동화함으로써 조직은 데이터 무결성을 향상하고 수동 오류를 줄이며 더 빠른 배치 출시를 달성할 수 있습니다.

로우 코드 애플리케이션 개발 플랫폼을 사용하면 조직은 시각적 도구와 최소한의 코딩을 사용하여 애플리케이션을 신속하게 설계, 구축 및 배포할 수 있습니다. 제약 산업에서는 데이터 무결성과 규제 표준을 유지하면서 더 빠른 혁신, 향상된 규정 준수, 효율적인 작업 흐름 자동화를 지원합니다.

로우코드 aPaaS(Application Platform as a Service) 플랫폼은 최소한의 코딩으로 애플리케이션을 빠르게 구축, 배포, 관리할 수 있는 클라우드 기반 환경을 제공합니다. 이를 통해 제약 조직은 워크플로를 간소화하고, 규정 준수를 보장하며, 운영을 효율적으로 확장하는 동시에 디지털 혁신을 가속화할 수 있습니다.

GMP SOP 교육 계획, 관리 및 추적 소프트웨어는 제약 회사가 규정 준수를 보장하면서 직원 교육 프로그램을 효율적으로 관리하는 데 도움이 됩니다. 교육 일정을 자동화하고, 진행 상황을 추적하고, 감사 준비 기록을 유지함으로써 조직은 인력 역량을 향상하고 규정 준수 프로세스를 간소화할 수 있습니다.

CAPA 관리 소프트웨어는 제약 회사가 시정 및 예방 조치를 효율적으로 관리하여 편차를 해결하고 품질 프로세스를 개선하도록 돕습니다. 워크플로를 자동화하고 추적성을 보장함으로써 규정 준수를 강화하고 위험을 줄이며 지속적인 개선을 지원합니다.

변경 제어 자동화 및 추적 소프트웨어는 제약 회사가 규정 준수를 보장하면서 변경 사항을 효율적으로 관리, 문서화 및 추적하는 데 도움이 됩니다. 워크플로우를 자동화하고 감사 추적을 유지함으로써 추적성을 향상시키고 위험을 줄이며 품질 관리 프로세스를 향상시킵니다.

QMS 자동화 소프트웨어는 제약 및 생명공학 기업이 품질 프로세스를 간소화하고 규정 준수를 관리하며 운영 효율성을 향상시키는 데 도움이 됩니다. CAPA, 편차, 감사, 문서 제어 등의 워크플로를 자동화하여 일관된 품질과 규정 준수를 보장합니다.

Exchange Server 관리 도구는 조직이 이메일 인프라를 효율적으로 관리, 모니터링 및 보호하는 데 도움이 됩니다. 관리 작업 자동화, 성능 추적, 보안 강화를 통해 안정적인 통신과 간소화된 IT 운영을 보장합니다.

코드 없는 애플리케이션 개발 플랫폼을 사용하면 사용자는 시각적 인터페이스와 사전 구축된 구성 요소를 사용하여 코드를 작성하지 않고도 애플리케이션을 구축하고 배포할 수 있습니다. 제약 산업에서는 더 빠른 혁신, 향상된 규정 준수 및 효율적인 작업 흐름 자동화를 가능하게 합니다.

Excel 자동화는 도구, 매크로 및 워크플로를 사용하여 반복적인 작업을 단순화하여 효율성과 정확성을 향상시킵니다. 제약 및 기업 환경의 경우 데이터 처리 속도를 높이고 수동 오류를 줄이며 효율적인 운영을 통해 생산성을 향상시킵니다.

환경 모니터링 시스템(EMS)은 제약 및 생명공학 시설에서 온도, 습도, 압력, 미생물 오염과 같은 중요한 환경 조건을 모니터링하는 데 필수적입니다. 실시간 데이터, 자동화된 경고 및 제조 환경 전반에 걸친 완벽한 추적성을 제공하여 제품 품질, 규정 준수 및 위험 감소를 보장합니다.

Quality Suite는 제약 및 생명공학 회사가 CAPA, 편차, 감사, 문서 제어 등의 품질 프로세스를 관리하는 데 도움이 되는 통합 플랫폼입니다. 워크플로우를 자동화하고 규정 준수를 보장함으로써 효율성, 데이터 무결성 및 전반적인 제품 품질을 향상시킵니다.

자산 관리 시스템은 조직이 조달에서 유지 관리 및 폐기에 이르기까지 자산의 수명주기를 추적, 관리 및 최적화하는 데 도움이 됩니다. 제약 및 기업 환경에서는 운영 효율성을 보장하고 가동 중지 시간을 줄이며 규정 준수를 지원합니다.

프로세스 검증 소프트웨어는 제약회사가 제조 프로세스에서 고품질 제품을 일관되게 생산하도록 보장합니다. 검증 작업 흐름을 자동화하고 상세한 문서를 유지함으로써 규정 준수를 강화하고 데이터 무결성을 향상하며 효율적인 수명 주기 관리를 지원합니다.

APQR 프로세스의 지연, 수동 스프레드시트 또는 추적성 부족으로 어려움을 겪고 계십니까? 이는 연간 제품 품질 검토가 규정을 준수할 준비가 되어 있지 않을 수 있다는 분명한 신호입니다. 상위 5개 경고 신호를 살펴보고 디지털 APQR 시스템이 어떻게 효율성을 개선하고 감사 준비를 보장하며 제약 운영의 규제 준수를 강화할 수 있는지 알아보세요.

APQR 프로세스의 비효율성으로 인해 어려움을 겪고 계십니까? 불완전한 데이터, 표준화 부족, 낮은 추적성과 같은 일반적인 실수는 규정 준수 및 의사 결정에 영향을 미칠 수 있습니다. 체계적인 디지털 접근 방식을 통해 이러한 함정을 피하고 연간 제품 품질 검토를 개선하는 방법을 알아보세요.

제약회사에서 감사 준비 상태를 유지하는 데 어려움을 겪고 계십니까? 수동 APQR 프로세스는 지연, 추적성 저하 및 규정 준수 위험으로 이어지는 경우가 많습니다. 디지털 APQR 시스템이 중앙 집중식 데이터, 실시간 가시성, 자동화된 문서화 및 완전한 감사 추적을 통해 감사 준비 상태를 어떻게 향상하는지 알아보세요.


















































FDA 3단계 CPV(지속적 공정 검증)는 제조 공정이 상업적 생산 중에 검증된 상태로 유지되도록 보장하는 공정 검증 수명주기의 중요한 단계입니다. 통계 도구와 실시간 데이터 분석을 사용하여 추세, 변동성 및 잠재적 편차를 감지하여 중요 공정 매개변수(CPP) 및 중요 품질 속성(CQA)을 지속적으로 모니터링하는 데 중점을 둡니다. FDA는 라이프사이클 접근 방식을 채택함으로써 검증이 일회성 활동이 아니라 데이터 및 위험 관리에 의해 주도되는 지속적인 프로세스임을 강조합니다. CPV는 프로세스 성능 데이터를 연간 제품 검토 및 관리 결정에 통합하여 사전 품질 보증 및 규정 준수를 지원합니다. AmpleLogic과 같은 고급 디지털 솔루션을 사용하면 조직은 CPV 구현을 간소화하고 데이터 무결성을 향상하며 진화하는 FDA 기대 사항을 지속적으로 준수할 수 있습니다.


21 CFR Part 11 준수 문서 관리 시스템은 제약 및 생명 과학 조직이 디지털 운영으로 전환하는 데 필수적입니다. 21 CFR Part 11은 전자 기록과 전자 서명이 신뢰할 수 있고 신뢰할 수 있으며 종이 기반 문서와 동등한 것으로 간주되는 기준을 설정합니다. 규정을 준수하는 문서 관리 시스템을 구현함으로써 조직은 완전한 감사 추적, 제어된 액세스 및 검증된 워크플로를 유지하면서 규제 대상 문서를 안전하게 생성, 저장, 검색 및 관리할 수 있습니다. 이러한 시스템은 추적 가능하고 조작할 수 없는 기록을 제공하여 데이터 무결성을 보장하고 무단 변경을 방지하며 규제 검사를 지원합니다. AmpleLogic과 같은 솔루션을 사용하면 기업은 문서 제어 프로세스를 디지털화하고, 전자 서명을 사용하여 승인을 자동화하고, FDA 규정을 완벽하게 준수할 수 있습니다. 이를 통해 궁극적으로 운영 효율성을 개선하고 규정 준수 위험을 줄이며 품질 및 규제 기능 전반에 걸쳐 감사 준비를 보장할 수 있습니다.

FDA 510(k) 허가 프로세스는 미국 시장에 진출하려는 의료기기 제조업체에게 중요한 규제 경로입니다. 시판 전 신고라고도 알려진 이 규정은 기업이 해당 기기가 안전성과 유효성 측면에서 이미 합법적으로 시판된 선행 기기와 "실질적으로 동등"함을 입증하도록 요구합니다. 클래스 II 및 일부 클래스 I 장치에 주로 적용되는 510(k) 프로세스에는 장치 설명, 용도, 라벨링, 위험 분석 및 성능 테스트 데이터를 포함한 자세한 문서 제출이 포함됩니다. FDA는 이 정보를 평가하여 기기가 통관 및 상업적 유통에 대한 규제 표준을 충족하는지 여부를 결정합니다. 제조업체는 AmpleLogic과 같은 디지털 및 자동화된 규정 준수 솔루션을 채택함으로써 문서 관리를 간소화하고, 데이터 무결성을 보장하고, 510(k) 제출 수명주기를 가속화할 수 있습니다. 이를 통해 규정 준수를 유지하면서 지연을 줄이고, 감사 준비를 개선하고, 혁신적인 의료 기기를 더 빠르게 시장에 출시할 수 있습니다.

실험실 계획 및 일정을 디지털화하는 것은 작업량 증가, 복잡한 테스트 요구 사항 및 엄격한 규정 준수 표준에 직면한 현대 QC 실험실에 필수적입니다. 종종 스프레드시트, 화이트보드 또는 기본 시스템에 의존하는 전통적인 접근 방식은 수천 개의 테스트를 효율적으로 조정해야 하는 실험실 운영의 복잡성이 증가하는 것을 관리하는 데 어려움을 겪습니다. 디지털 솔루션을 채택함으로써 실험실은 효율성을 크게 향상시키고 처리 시간을 단축하며 자원 활용도를 최적화할 수 있습니다. 입증된 이점에는 더 빠른 테스트 실행, 재고 요구 사항 감소, 우선 순위 기반 테스트 워크플로 준수 향상 등이 포함됩니다. 주요 모범 사례에는 시뮬레이션을 위한 디지털 트윈 모델링 구현, 중요 경로 테스트 우선 순위 지정, 캠페인 기반 실행 활용, 리소스 계획 통합, 실시간 적응형 일정 지원 등이 포함됩니다. 이러한 전략은 더 나은 가시성을 제공하고 병목 현상을 최소화하며 의사 결정을 향상시킵니다. AmpleLogic과 같은 플랫폼을 사용하면 조직은 지능형 일정 관리, AI 기반 통찰력 및 실시간 모니터링을 통합하여 실험실 운영을 혁신하여 규정 준수를 보장하고 생산성을 향상시키며 생명 과학 환경의 지속적인 개선을 위한 확장 가능한 기반을 구축할 수 있습니다.

배치 출시는 의약품 제조에서 가장 중요하고 규제가 엄격한 단계 중 하나이며 제품 배포 전에 모든 생산 및 품질 데이터를 철저히 검토하고 승인해야 합니다. 기존의 수동 검토 프로세스는 시간이 많이 걸리고 오류가 발생하기 쉽고 추적하기 어려워 지연 및 규정 준수 위험이 발생하는 경우가 많습니다. AmpleLogic의 PQR 소프트웨어는 전체 검토 프로세스를 표준화하고 간소화하는 자동화된 배치 릴리스 체크리스트를 도입하여 이러한 문제를 해결합니다. BMR(배치 제조 기록), 편차, CAPA 및 품질 시스템의 데이터를 체계적이고 감사 가능한 워크플로우로 통합하여 모든 단계에서 완전성과 추적성을 보장합니다. 구성 가능한 SOP 기반 체크리스트, 자동화된 검증 플래그, 디지털 서명 및 실시간 대시보드를 갖춘 이 플랫폼을 통해 QA 팀은 문제를 조기에 감지하고 수동 작업을 줄이며 배치 승인을 가속화할 수 있습니다. 그 결과 데이터 무결성이 향상되고 규정 준수가 향상되며 출시 기간이 단축되어 일괄 릴리스가 더욱 효율적이고 안정적이며 검사 준비가 완료됩니다.

FDA 경고장은 제약회사가 제품 품질과 환자 안전을 보장하는 데 중요한 CGMP(현행 우수제조관리기준)와 같은 규제 표준을 준수하지 못할 때 발행됩니다. 이러한 서신은 사전에 해결하지 않을 경우 제품 리콜, 운영 중단, 평판 손상으로 이어질 수 있는 반복적인 규정 준수 격차를 강조하는 경우가 많습니다. FDA 경고 서한에서 확인된 일반적인 문제로는 부적절한 원자재 테스트, 공급업체 자격 부족, 적절한 문서화 및 품질 감독 부족, 공정 및 세척 검증 부족, 실험실 제어 및 데이터 무결성 실패 등이 있습니다. 또한 안정성 테스트, 배치 기록 관리, 환경 모니터링, 변경 제어 프로세스의 결함이 규제 기관에서 자주 언급됩니다. 이러한 과제를 해결하기 위해 조직에서는 추적성을 보장하고 규정 준수를 강화하며 인적 오류를 줄이는 디지털 품질 관리 시스템, LIMS 및 자동화된 워크플로를 점점 더 많이 채택하고 있습니다. AmpleLogic이 제공하는 것과 같은 통합 디지털 솔루션을 구현함으로써 기업은 위험을 사전에 식별하고, 품질 프로세스를 간소화하고, 지속적인 감사 준비 상태를 유지할 수 있습니다. 결과적으로 비용이 많이 드는 FDA 경고 서신을 피하고 장기적인 규정 준수를 보장할 수 있습니다.

식음료 실험실은 생산 라이프사이클 전반에 걸쳐 제품 안전, 품질 및 규정 준수를 보장하는 데 중요한 역할을 합니다. 원자재 테스트부터 완제품 검증까지 이러한 실험실에서는 복잡한 작업 흐름, 대용량 데이터, 엄격한 식품 안전 표준을 관리해야 합니다. 식품 및 음료 실험실용 LIMS 소프트웨어는 샘플 추적을 간소화하고, 데이터 캡처를 자동화하고, ISO 17025, HACCP, 식품 안전 표준과 같은 글로벌 규정 준수를 보장하는 중앙 집중식 플랫폼을 제공합니다. 재료 조달부터 최종 제품 출시까지 엔드투엔드 추적성을 지원하는 동시에 수동 오류를 줄이고 데이터 무결성을 향상시킵니다. 배치 및 로트 추적성, 장비 통합, 자동화된 작업 흐름, 실시간 보고 등의 고급 기능을 갖춘 최신 LIMS 솔루션은 실험실에서 운영 효율성을 향상시키고 테스트 주기를 가속화하며 감사 준비 상태를 유지할 수 있도록 지원합니다. AmpleLogic과 같은 플랫폼을 채택함으로써 조직은 식품 품질 관리 프로세스를 혁신하고 일관되고 규정을 준수하는 고품질 제품 결과를 보장할 수 있습니다.

제약 및 생명 과학 조직이 증가하는 규제 압력과 운영 복잡성에 직면함에 따라 종이 기반 QMS와 디지털 QMS 간의 논쟁이 그 어느 때보다 중요해졌습니다. 한때 표준이었던 기존의 종이 기반 시스템은 이제 수동 문서화, 느린 승인, 감사 준비 유지의 어려움 등의 비효율성 문제로 어려움을 겪고 있습니다. 종이 기반 QMS는 문서 위치 오류, 버전 관리 문제, 품질 프로세스에 대한 가시성 제한 등의 위험을 초래하는 경우가 많아 규제가 엄격한 환경에서는 규정 준수 관리가 더욱 어려워집니다. 이와 대조적으로 디지털 QMS(eQMS)는 모든 품질 프로세스를 중앙 집중화하고 작업 흐름을 자동화하며 정확하고 추적 가능한 데이터에 대한 실시간 액세스를 보장합니다. 디지털 QMS 솔루션은 자동화된 문서 제어, 보안 감사 추적, 전자 서명, 실시간 보고 대시보드 등 상당한 이점을 제공합니다. 이러한 기능은 FDA 21 CFR Part 11 및 GMP 표준과 같은 규정을 준수하는 동시에 데이터 무결성을 강화하고 인적 오류를 줄이며 전반적인 운영 효율성을 향상시킵니다. AmpleLogic과 같은 플랫폼을 사용하면 조직은 사후 대응적인 수동 품질 관리에서 사전 예방적인 데이터 중심 접근 방식으로 전환하여 더 빠른 의사 결정, 향상된 규정 준수 및 확장 가능한 품질 운영을 가능하게 할 수 있습니다. 오늘날의 디지털 시대에 최신 QMS는 단순한 업그레이드가 아니라 경쟁력과 규제 우수성을 유지하기 위한 전략적 필요성입니다.

생명 과학 산업에서 문서 관리는 엄격한 규제 요구 사항, 높은 데이터 볼륨, 완벽한 추적성 요구로 인해 복잡합니다. 규정 준수 문서 관리 시스템(DMS)은 조직이 FDA 21 CFR Part 11, EU Annex 11 및 GxP 지침과 같은 글로벌 규정을 준수하도록 보장하면서 문서 프로세스를 디지털화, 제어 및 표준화하는 데 도움이 됩니다. 기존의 종이 기반 시스템이나 단편화된 시스템은 종종 비효율성, 버전 제어 문제 및 규정 준수 위험을 초래합니다. 이와 대조적으로 최신 전자 문서 관리 시스템은 작성 및 검토부터 승인, 릴리스 및 보관에 이르기까지 문서 수명주기에 대한 중앙 집중식 제어를 제공하여 정확성, 일관성 및 감사 준비를 보장합니다. 전자 서명, 감사 추적, 자동화된 워크플로, AI 기반 검색과 같은 고급 기능을 갖춘 AmpleLogic과 같은 플랫폼을 통해 생명 과학 조직은 데이터 무결성을 향상하고 협업을 강화하며 수동 작업을 줄일 수 있습니다. 문서 제어 프로세스를 디지털화함으로써 기업은 더 빠른 승인, 더 나은 규정 준수 가시성, 규제가 심한 환경에서 규제 우수성을 위한 확장 가능한 기반을 확보할 수 있습니다.

제약회사는 규정 준수, 혁신, 운영 효율성을 유지하면서 IT 비용을 절감해야 한다는 점점 더 큰 압력을 받고 있습니다. 증가하는 인프라 비용, 기존 시스템 및 복잡한 규제 요구 사항으로 인해 IT는 업계에서 가장 중요한 비용 센터 중 하나가 되었습니다. 전략적 IT 비용 절감은 시스템 효율성 향상, 중복 제거, 최신 기술 활용을 통해 비용을 절감하는 것뿐만 아니라 최적화하는 데 중점을 둡니다. 예를 들어, IT 인프라 통합, 애플리케이션 합리화, 지원 서비스 아웃소싱을 통해 운영 오버헤드를 크게 줄일 수 있습니다. 연구에 따르면 IT 시스템과 공급업체 관리를 합리화하는 것만으로도 단기간에 총 IT 비용을 약 30% 절감할 수 있는 것으로 나타났습니다. 주요 전략에는 클라우드 기반 플랫폼 채택, 수동 프로세스 자동화, 품질 및 규정 준수 시스템 통합, 단절된 여러 도구에 대한 의존도 감소 등이 포함됩니다. 또한 전문 기능을 아웃소싱하고 리소스 활용도를 개선하면 효율성이 크게 향상될 수 있으며 일부 조직에서는 이러한 결합된 접근 방식을 통해 극적인 비용 절감을 달성할 수 있습니다. AmpleLogic과 같은 플랫폼을 통해 제약 회사는 시스템을 통합하고, 워크플로를 자동화하고, 데이터 가시성을 향상시켜 보다 스마트한 의사 결정과 확장 가능한 디지털 혁신을 실현할 수 있습니다. 자동화, 통합 및 프로세스 최적화의 올바른 조합을 구현함으로써 조직은 IT 비용을 크게 낮추는 동시에 규정 준수, 민첩성 및 전반적인 비즈니스 성과를 향상시킬 수 있습니다.

효과적인 APQR(연간 제품 품질 검토)을 수행하는 것은 제약 산업에서 일관된 제품 품질과 규정 준수를 보장하는 데 중요합니다. 그러나 조직은 프로세스를 복잡하고 시간이 많이 걸리며 오류가 발생하기 쉬운 여러 가지 문제에 직면하는 경우가 많습니다. 주요 과제 중 하나는 LIMS, QMS, ERP, 스프레드시트 등의 시스템에 분산된 데이터가 단편화되어 데이터 집계가 어렵고 오류가 발생하기 쉽다는 것입니다. 또한 수동으로 데이터를 처리하면 불일치, 중복 및 데이터 무결성 문제가 발생하여 APQR 보고서의 정확성이 손상될 수 있습니다. 기타 일반적인 문제로는 규정 준수 압력, 추세 분석을 위한 제한된 분석 기능, 비효율적인 부서간 협업, 감사 중 추적성 부족 등이 있습니다. 이러한 문제로 인해 검토 프로세스가 지연되고 수동 작업이 증가하며 효율성이 저하되는 경우가 많습니다. 이러한 과제를 극복하기 위해 제약 회사는 데이터 소스를 통합하고 워크플로를 자동화하며 실시간 분석을 제공하는 디지털 솔루션을 채택하고 있습니다. 표준화된 SOP를 구현하고, 데이터 거버넌스를 개선하고, 부서 간 협업을 활성화하고, 고급 APQR 소프트웨어를 활용하면 효율성이 크게 향상되고, 규정 준수가 보장되며, APQR을 사전 예방적인 데이터 중심 품질 프로세스로 전환할 수 있습니다.

의료기기로서의 소프트웨어(SaMD)는 물리적 의료기기의 일부가 아닌 진단, 치료, 모니터링 등의 의료 기능을 수행하기 위한 소프트웨어를 의미합니다. IMDRF(국제의료기기규제자포럼) 및 FDA와 같은 글로벌 규제 기관에 따르면 SaMD는 컴퓨터, 모바일 장치 또는 클라우드 환경과 같은 범용 플랫폼에서 작동하면서 임상적으로 관련 있는 결과를 제공합니다. SaMD 솔루션은 진단 영상 분석 및 임상 결정 지원 시스템부터 환자 상태를 모니터링하는 모바일 건강 애플리케이션에 이르기까지 의료 전반에 걸쳐 널리 사용됩니다. 이러한 시스템은 환자의 안전과 효율성을 보장하기 위해 위험 분류, 검증, 사이버 보안, 데이터 무결성 표준을 포함한 엄격한 규제 요구 사항을 준수해야 합니다. 디지털 건강 및 AI 기반 기술의 급속한 성장으로 SaMD는 더 빠른 진단, 맞춤형 치료 및 실시간 모니터링을 지원함으로써 의료 서비스 제공 방식을 변화시키고 있습니다. 그러나 규정 준수, 품질 보증, 수명 주기 관리와 관련된 문제도 발생합니다. AmpleLogic과 같은 플랫폼은 조직이 검증을 간소화하고, 규정 준수를 보장하고, 의료 기기 소프트웨어의 전체 수명주기를 관리하도록 지원하여 안전 및 규정 준수 표준을 유지하면서 더 빠른 혁신을 가능하게 합니다.

의료 기기에 대한 품질 관리 시스템(QMS)은 설계 및 개발부터 제조, 시판 후 감시에 이르기까지 전체 수명주기 동안 제품 품질, 안전 및 규정 준수를 보장하도록 설계된 프로세스, 절차 및 책임의 구조화된 프레임워크입니다. 의료 기기 제조업체는 일관된 제품 품질과 환자 안전을 보장하기 위해 국제 표준에 부합하는 ISO 13485, EU MDR 및 FDA의 품질 관리 시스템 규정(QMSR)과 같은 엄격한 글로벌 규정을 준수해야 합니다. 강력한 QMS는 설계 제어, 위험 관리, 공급업체 품질 관리, CAPA(시정 및 예방 조치), 교육 관리, 감사 프로세스 등의 중요한 요소를 통합합니다. 이러한 구성 요소는 조직이 추적성을 유지하고 위험을 최소화하며 운영 전반에 걸쳐 지속적인 개선을 보장하는 데 도움이 됩니다. 의료기기 회사는 AmpleLogic과 같은 디지털 QMS 솔루션을 채택함으로써 품질 프로세스를 간소화하고 규정 준수 워크플로우를 자동화하며 데이터 무결성을 향상시킬 수 있습니다. 이를 통해 규제가 엄격한 산업에서 더 빠른 규제 승인, 강화된 감사 준비, 안전한 고품질 의료 기기의 일관된 제공이 가능해집니다.

인공 지능은 기존 수동 프로세스의 비효율성을 해결하여 제약 및 생명과학 산업의 품질 관리 시스템(QMS)을 빠르게 변화시키고 있습니다. 기존 QMS는 단편화된 데이터, 지연된 조사, 품질 이벤트에 대한 제한된 가시성으로 인해 규정 준수 및 운영 효율성에 영향을 미치는 경우가 많습니다. AI를 QMS에 통합함으로써 조직은 CAPA, 편차 관리, 변경 제어 및 감사 관리와 같은 중요한 품질 프로세스를 자동화할 수 있습니다. AI 기반 시스템은 기계 학습과 예측 분석을 활용하여 추세를 식별하고, 이상 징후를 감지하고, 시정 조치를 권장하므로 근본 원인을 더 빠르게 분석하고 사전에 위험을 완화할 수 있습니다. AI 기반 QMS는 또한 실시간 통찰력을 제공하고 데이터 정확성을 향상하며 수동 개입을 줄여 의사결정을 향상시킵니다. 예측적 OOS/OOT 감지, 자동화된 불만 처리, 지능형 위험 평가와 같은 기능은 조직이 제품 품질을 개선하고 규정 준수를 보장하며 운영 비용을 절감하는 데 도움이 됩니다. AmpleLogic과 같은 플랫폼을 통해 제약 회사는 완벽하게 통합된 AI 기반 품질 생태계를 채택하여 워크플로를 간소화하고, 승인을 가속화하며, 상당한 효율성 향상을 달성하는 동시에 글로벌 규정을 엄격하게 준수할 수 있습니다.

의료 기기 문서 제어는 설계, 제조, 품질 관리와 관련된 모든 문서가 라이프사이클 전반에 걸쳐 적절하게 생성, 검토, 승인 및 유지되도록 보장하는 품질 및 규정 준수의 중요한 구성 요소입니다. 이는 규제가 엄격한 환경에서 제품 안전, 추적성 및 감사 준비 상태를 유지하는 데 중요한 역할을 합니다. FDA 21 CFR Part 820, ISO 13485 및 EU MDR과 같은 규제 프레임워크에서는 조직이 문서 승인, 버전 제어, 배포 및 변경 관리를 위한 공식 절차를 확립하도록 요구합니다. 이러한 규정은 승인된 최신 문서만 사용하고 모든 변경 사항을 추적하고 검증하여 데이터 무결성을 유지하도록 보장합니다. 강력한 문서 제어 시스템을 통해 조직은 DMR(장치 마스터 기록), DHR(장치 기록 기록) 및 품질 절차와 같은 중요한 기록을 완전한 추적성을 통해 관리할 수 있습니다. AmpleLogic과 같은 디지털 솔루션을 채택함으로써 기업은 문서 워크플로우를 자동화하고, 규정 준수를 강화하고, 안전한 감사 추적을 유지하고, 운영 효율성을 향상시켜 규제 검사 및 장기적인 품질 우수성에 대한 준비를 보장할 수 있습니다.

품질 핵심 성과 지표(KPI)는 의약품 제조에서 품질 관리 시스템(QMS)의 효율성을 평가하는 데 사용되는 필수 지표입니다. 이러한 KPI는 제품 품질, 프로세스 효율성 및 규정 준수에 대한 측정 가능한 통찰력을 제공하여 조직이 데이터 기반 결정을 내리고 지속적인 개선을 보장하도록 돕습니다. 제약 제조에서 일반적으로 추적되는 KPI에는 배치 거부율, 편차율, CAPA 효율성, OOS(사양 이탈) 사고 및 조사 주기 시간이 포함됩니다. 이러한 지표는 프로세스 비효율성을 식별하고, 품질 문제를 조기에 감지하며, 적시에 시정 조치를 취하는 데 도움이 됩니다. 이러한 KPI를 모니터링하는 것은 GMP 표준 준수를 유지하고 일관된 제품 품질을 보장하는 데 중요합니다. 또한 RFT(Right First Time), 결함률 및 프로세스 주기 시간과 같은 지표는 제조 성능 및 운영 효율성에 대한 통찰력을 제공합니다. 이러한 KPI를 분석함으로써 조직은 재작업을 줄이고 비용을 최소화하며 전반적인 생산 결과를 향상시킬 수 있습니다. 제약회사는 AmpleLogic과 같은 디지털 플랫폼을 사용하여 KPI 추적을 자동화하고 실시간 대시보드를 생성하며 예측 통찰력을 얻을 수 있습니다. 이를 통해 품질 관리를 규정 준수, 효율성 및 제품 신뢰성을 향상시키는 사전 예방적인 데이터 기반 기능으로 전환할 수 있습니다.

안정성 연구는 의약품 개발의 중요한 구성 요소로, 의약품이 정의된 환경 조건에서 유통기한 동안 정체성, 강도, 품질 및 순도를 유지하도록 보장합니다. 이러한 연구는 ICH 지침에 따라 만료 날짜, 보관 요구 사항 및 규정 준수를 확립하는 데 도움이 됩니다. 회귀 분석은 성능 저하 패턴을 모델링하고 제품 유효 기간을 예측하여 안정성 데이터를 해석하는 데 중요한 역할을 합니다. 회귀 기법은 온도, 습도, 빛 노출 등의 요소에 영향을 받는 추세를 분석하여 유통기한을 정확하게 추정하고 제약 품질 관리에서 데이터 기반 의사결정을 지원합니다. ANOVA 및 ANCOVA와 같은 고급 통계 방법은 조건에 따른 중요한 변화를 식별하고 외부 변수를 조정하여 안정성 분석을 더욱 향상시킵니다. 이러한 접근 방식은 안정성 예측의 정확성과 신뢰성을 향상시켜 일관된 제품 성능과 규정 준수를 보장합니다. AmpleLogic의 안정성 연구 관리 소프트웨어와 같은 디지털 솔루션을 활용함으로써 조직은 워크플로를 자동화하고, 실시간 추세를 모니터링하고, 지능형 통계 모델을 적용하여 안정성 테스트를 보다 효율적이고 정확하며 규정을 준수하는 프로세스로 전환하여 장기적인 의약품 신뢰성을 보장할 수 있습니다.

편차 관리는 규제 표준에 따라 모든 부적합 사항을 적절하게 식별, 조사 및 해결하도록 보장하는 제약 품질 시스템의 중요한 구성 요소입니다. 그러나 기존 접근 방식은 느리고 수동적이며 리소스 집약적인 경우가 많아 조사가 지연되고 규정 준수 위험이 증가하는 경우가 많습니다. 인공 지능은 데이터 수집, 근본 원인 분석, CAPA 권장 사항과 같은 주요 단계를 자동화하여 편차 관리를 혁신하고 있습니다. AI 기반 시스템은 과거 편차를 분석하고, 프로세스와 장비 전반의 패턴을 식별하며, 품질 팀이 더 빠르고 정확한 결정을 내리는 데 도움이 되는 데이터 기반 통찰력을 제공합니다. AmpleLogic의 QMS와 같은 AI 지원 솔루션을 사용하면 조직은 조사 일정을 크게 단축하고 CAPA 효율성을 개선하며 편차 재발을 최소화할 수 있습니다. 이러한 시스템은 추적성을 향상시키고, 감사 준비를 보장하며, 사전 대응적인 문제 처리에서 예측 가능한 인텔리전스 기반 규정 준수로 전환하여 사전 품질 관리를 지원합니다.

안정성 소프트웨어는 의약품 개발 라이프사이클 전반에 걸쳐 규정 준수를 보장하고 제품 품질을 유지하는 데 중요한 역할을 합니다. 안정성 연구는 다양한 환경 조건에서 의약품이 어떻게 정체성, 강도, 품질 및 순도를 유지하는지 결정하는 데 필수적이며 규제 승인 및 수명 주기 관리의 초석이 됩니다. 최신 안정성 관리 소프트웨어를 통해 조직은 ICH 준수 안정성 연구를 효율적으로 설계, 실행 및 모니터링할 수 있습니다. 이러한 시스템은 프로토콜 관리, 시료 채취 일정 관리, 환경 모니터링, 통계 추세 분석 등의 중요한 프로세스를 자동화하여 정확한 유통기한 예측과 강력한 규제 제출을 보장합니다. 안정성 워크플로를 디지털화함으로써 제약 회사는 수동 추적을 제거하고, 데이터 무결성을 개선하며, FDA 21 CFR Part 11 및 GxP 요구 사항에 부합하는 완전한 감사 추적을 유지할 수 있습니다. 또한 고급 플랫폼은 LIMS 및 QMS 시스템과 통합되어 연구 진행 상황에 대한 실시간 가시성을 제공하고 사전 예방적인 의사 결정을 가능하게 합니다. AmpleLogic과 같은 솔루션을 사용하면 조직은 안정성 연구 관리를 간소화하고 규정 준수를 강화하며 일관된 제품 품질을 보장하여 궁극적으로 규제가 엄격한 환경에서 개발 일정을 가속화하고 전반적인 운영 효율성을 향상시킬 수 있습니다.























제약 분야의 APQR(연간 제품 품질 검토)은 제품 품질, 제조 일관성, 편차 및 시간 경과에 따른 규정 준수를 평가하여 지속적인 개선과 규제 준수를 보장하는 GMP 요구 사항입니다.

제약 분야 APQR(제품 품질 검토)의 주요 과제에는 데이터 통합 문제, 일관되지 않은 문서화, 편차 관리, 규제 준수 격차, 제한된 추세 분석 등이 포함되어 품질, 효율성 및 지속적인 개선에 영향을 미칩니다.

제약 분야의 수동 및 자동 APQR은 효율성, 데이터 정확성, 규정 준수 및 확장성의 주요 차이점을 탐색하여 제약 업계 리더가 품질 검토 및 지속적인 개선을 위한 올바른 접근 방식을 선택하도록 돕습니다.

Gartner의 실험실 정보 관리 시스템(LIMS) 시장 가이드에 AmpleLogic이 포함된 것은 생명 과학 기술 분야에서 AmpleLogic의 영향력이 커지고 있음을 강조합니다. 로우 코드 AI 기반 플랫폼을 갖춘 AmpleLogic은 실험실에서 워크플로를 간소화하고 데이터 무결성을 향상하며 디지털 혁신 이니셔티브를 가속화할 수 있도록 지원합니다. 이러한 인정은 현대 실험실을 위한 혁신적이고 규정을 준수하며 확장 가능한 솔루션을 제공하려는 회사의 노력을 반영합니다.

APQR 프로세스 내에서 배치 릴리스를 자동화하면 제약 회사가 품질 및 규정 준수를 관리하는 방식이 변화됩니다. 전통적으로 배치 출시에는 제품이 유통에 적합한지 확인하기 위해 제조 및 품질 기록에 대한 광범위한 수동 검토가 포함됩니다. APQR(연간 제품 품질 검토)은 1년 동안 모든 배치의 데이터를 통합하여 추세를 파악하고 일관된 제품 품질을 보장합니다. 자동화를 APQR에 통합함으로써 조직은 배치 검토 워크플로를 디지털화하고 체크리스트 기반 검증을 구현하며 편차, CAPA 및 품질 지표를 실시간으로 추적할 수 있습니다. 이를 통해 데이터 무결성과 감사 준비가 향상될 뿐만 아니라 릴리스 주기 시간도 크게 단축됩니다. AmpleLogic과 같은 플랫폼을 사용하면 제약 회사는 대응적인 품질 검사에서 사전 예방적인 데이터 기반 배치 출시 결정으로 전환하여 규정 준수를 저해하지 않고 더 빠른 승인을 보장할 수 있습니다.














분석 테스트에서 사양 이탈(OOS) 결과를 조사하기 위한 포괄적인 가이드입니다. 규제 대상 산업에서 데이터 무결성, 품질 보증 및 효과적인 CAPA 관리를 보장하기 위해 AmpleLogic의 eQMS를 사용하여 구조화된 워크플로, 근본 원인 분석 및 규정 준수 접근 방식을 알아보세요.

AmpleLogic의 eQMS가 제약 산업에서 품질 관리 시스템의 수명주기를 어떻게 단순화하는지 알아보세요. 확장 가능한 고급 플랫폼을 사용하여 배포부터 지속적인 개선까지 원활한 규정 준수를 달성하고, 제품 품질을 개선하고, 중요한 품질 프로세스를 디지털화하세요.

생명 과학 분야에서 인적 오류는 규정 준수 위험과 값비싼 편차로 이어질 수 있습니다. 워크플로를 간소화하고, 데이터 캡처를 자동화하고, 제약 및 생명공학 운영 전반에서 정확성을 보장하는 6가지 필수 디지털 도구를 살펴보세요.

인적 오류는 여전히 사이버 보안 침해의 주요 원인 중 하나로 남아 있으며, 산업 전반에 걸쳐 사고의 최대 95%를 차지합니다. 생명 과학에서는 사소한 실수라도 민감한 데이터를 손상시키고 규정 준수를 방해하며 환자 안전에 영향을 미칠 수 있으므로 강력한 디지털 시스템이 필수적입니다.

Cipla가 AmpleLogic의 고급 UAM 솔루션을 사용하여 제약 운영에서 사용자 액세스 관리를 어떻게 간소화했는지 알아보세요. 이 사례 연구에서는 개선된 규정 준수, 자동화된 사용자 프로비저닝, GxP 요구 사항에 부합하는 안전하고 감사 가능한 액세스 제어를 강조합니다.

의약품 제조에서 MACO 계산 및 세척 검증에 대한 규제 요구 사항을 이해합니다. 이 가이드에서는 FDA, EMA, CDSCO, WHO와 같은 글로벌 기관이 규정 준수, 환자 안전 및 감사 준비를 보장하기 위해 PDE 및 용량 기반 접근 방식을 사용하여 과학적으로 정당한 위험 기반 잔류 제한을 기대하는 방법을 설명합니다.

인적 오류는 의약품 제조에서 규정 준수 문제의 주요 원인 중 하나로 남아 있습니다. eBMR, QMS, LIMS와 같은 디지털 솔루션을 채택함으로써 조직은 수동 비효율성을 제거하고 데이터 정확성을 향상시키며 자동화, 실시간 모니터링 및 표준화된 워크플로를 통해 규정 준수를 보장할 수 있습니다.

Teva가 어떻게 AmpleLogic의 eQMS를 활용하여 품질 프로세스를 디지털화하고 규정 준수를 개선하며 수동 작업 부하를 줄이는지 알아보세요. 이 사례 연구에서는 통합 디지털 플랫폼을 통해 제약 산업에서 어떻게 더 빠른 운영, 더 나은 감사 준비, 확장 가능한 품질 관리가 가능했는지 강조합니다.

오래된 애플리케이션 소프트웨어는 조직을 심각한 보안 위험, 규정 준수 실패 및 운영 비효율성에 노출시킬 수 있습니다. 이 기사에서는 레거시 시스템이 제약과 같은 규제 산업에 어떤 영향을 미치는지, 그리고 현대적이고 안전한 디지털 솔루션으로의 업그레이드가 필수적인 이유를 살펴봅니다. AmpleLogic이 기업의 규정 준수, 보안 및 미래 대비를 유지하는 데 어떻게 도움이 되는지 알아보세요.

화장품 산업을 위한 AmpleLogic의 LMS는 교육을 간소화하고 규정 준수를 보장하며 인력 역량을 향상시킵니다. AI 기반 자동화, SOP 관리 및 GMP 지원 기능을 통해 화장품 회사는 규제가 엄격한 환경에서 품질, 안전 및 감사 준비 상태를 유지할 수 있습니다.

FDA 소프트웨어 검증 프로세스는 제약 분야의 컴퓨터 시스템이 규제 요구 사항을 충족하면서 의도한 대로 일관되게 작동하는지 확인하는 데 필수적입니다. 이 블로그에서는 검증 수명주기 단계, 문서화, 위험 기반 접근 방식과 디지털 도구가 규정 준수, 데이터 무결성 및 감사 준비를 달성하는 데 어떻게 도움이 되는지 설명합니다.

AmpleLogic의 QC 계획 및 예약 소프트웨어는 지능형 계획, 자동화된 예약 및 캠페인 기반 실행을 통해 제약 실험실을 지원합니다. 자원 활용도를 향상시키고 처리 시간을 단축하며 실시간 가시성과 최적화된 실험실 워크플로를 통해 규정 준수를 보장합니다.

실용적인 eLogbook 체크리스트는 QA, QC 및 생산 팀이 문서를 표준화하고 인적 오류를 줄이며 GMP 규정 준수를 유지하는 데 도움이 됩니다. 제약회사는 로그북을 디지털화함으로써 데이터 무결성을 향상하고 감사 준비를 보장하며 일일 운영 워크플로를 간소화할 수 있습니다.

OCuSOFT가 어떻게 AmpleLogic의 eQMS를 활용하여 엔드 투 엔드 품질 프로세스를 디지털화하고 종이 기반 기록을 85% 줄이면서 가시성, 규정 준수 및 감사 준비 상태를 개선했는지 알아보세요. 이 솔루션은 규제된 환경에서 문서 제어, CAPA 및 감사 워크플로를 간소화했습니다.

FDA 483 관찰 내용에 대응하려면 시기 적절하고 체계적이며 증거 기반 접근 방식이 필요합니다. 조직은 경고 편지를 피하고 규정 준수를 강화하기 위해 근본 원인 분석, 시정 및 예방 조치(CAPA), 명확한 일정을 통해 각 관찰 사항을 해결해야 합니다.

시설 변경에서 QA를 제외하면 규정 준수 격차, 검증 실패 및 규제 위험 증가로 이어질 수 있습니다. QA는 구조화된 변경 제어 및 위험 관리 프로세스를 통해 영향을 평가하고, 적절한 문서화를 보장하고, GMP 표준을 유지하는 데 중요한 역할을 합니다.

의약품 배치 발행 절차는 GMP 준수, 추적성 및 제품 품질을 보장하는 데 중요합니다. BMR(배치 제조 기록) 발행부터 원자재 확인 및 모든 단계 문서화에 이르기까지 이러한 프로세스는 각 배치에 대한 완전한 감사 추적을 제공하고 규제 검사를 지원합니다.

불완전한 배치 기록은 FDA 감사 중 주요 위험 신호로, 종종 483건의 관찰 및 경고 서한으로 이어집니다. 서명 누락, 불분명한 항목 및 데이터 격차로 인해 제품 품질, 추적성 및 데이터 무결성에 대한 우려가 높아지므로 규정 준수를 위해 강력한 문서화 관행이 필수적입니다.
















ADC Therapeutics가 AmpleLogic을 통해 표준 관리를 디지털화하여 규정 준수를 개선하고 수작업을 줄이며 데이터 가시성과 효율성을 향상한 방법을 알아보세요.

제약 업계에서 흔히 발생하는 MHRA 데이터 무결성 오류와 이를 방지하는 방법을 알아보세요. 규정 준수를 개선하고, ALCOA+ 원칙을 보장하며, 모범 사례를 통해 감사 준비 상태를 유지하세요.

멸균 LIMS가 제약 분야의 OSD LIMS와 어떻게 다른지 알아보세요. 워크플로, 규정 준수 요구 사항, 데이터 관리를 비교하여 올바른 솔루션을 선택하세요.

인간의 실수가 제약 업계의 데이터 무결성에 어떤 영향을 미치는지 이해합니다. FDA 규정 준수 요구 사항, ALCOA+ 원칙 및 감사 실패를 방지하기 위한 입증된 전략에 대해 알아보세요.

분석 테스트에서 OOS(사양 이탈) 결과를 조사하는 방법을 알아보세요. 제약 규정 준수를 위한 근본 원인 분석, FDA 지침 및 CAPA 전략을 이해합니다.


















AI와 코드 없는 플랫폼이 더욱 스마트한 품질 관리, 예측 유지 관리, 검사 준비 문서를 통해 제약 제조를 어떻게 변화시키고 있는지 알아보세요.

Discover the hidden operational debt pharma companies face by delaying low-code and AI adoption—and how AmpleLogic helps reduce it with GxP-compliant workflows.

AmpleLogic으로 연간 제품 품질 검토를 자동화하세요. 준비 시간을 70~80% 줄이고, GMP 규정 준수를 보장하며, 규제에 맞는 PQR 보고서를 더 빠르게 생성하세요.

제약 규제 팀이 AmpleLogic RIMS를 사용하여 제출, 갱신, CMC 업데이트 및 글로벌 파일링을 정확하게 관리하기 위해 RIMS 솔루션이 필요한 이유를 알아보세요.

AI 기반 솔루션으로 규정 준수를 보장하고, 수동 오류를 제거하고, 데이터 무결성을 개선하기 위해 제약 공장에 eLogbook이 필요한 이유를 알아보세요.

AmpleLogic AI는 의약품 편차 처리 시간을 줄이고 근본 원인 분석을 자동화하며 규정 준수를 개선하고 조사 속도를 높입니다.

제약 분야의 세척 검증에 대한 AmpleLogic 가이드는 GMP 준수를 보장하고 오염을 방지하며 제조 효율성을 향상시킵니다.


Eisai Pharmaceuticals는 AmpleLogic 직원 교육 관리 소프트웨어를 사용하여 성공적으로 사업을 시작했습니다.

Hetero Labs Limited, AmpleLogic 교정 및 예방 유지 관리 소프트웨어 출시













제약 제조업체로서 귀하는 업계에 적용되는 엄격한 규제 요건을 잘 알고 있을 것입니다. 규정 준수의 중요한 측면 중 하나는 정확한 최신 기록과 문서를 유지하는 것입니다. 하지만 기록 보관 시스템에서 일지의 역할을 고려해 보셨나요? 그렇지 않은 경우 시작하는 것이 좋습니다.

최근 생명과학 산업에 디지털 기술을 접목하는 것은 비즈니스 필수 사항이 되었습니다. 일일 규제 업데이트, 증가하는 문서화, 보다 빠른 감사의 필요성으로 인해 제약회사는 수동 프로세스에서 꾸준히 벗어나고 있습니다. 그러한 변화 중 하나는 규정 준수, 효율성 및 데이터 무결성을 보장하는 데 점점 더 중요해지고 있는 도구인 문서 관리 시스템(DMS)으로의 전환입니다. 디지털 혁신은 모든 부문에 영향을 미쳤지만 제약 운영 및 품질 보증에서 디지털 혁신의 역할은 특히 혁신적입니다. 업계를 변화시키는 많은 도구 중에서 DMS는 문서 작업 흐름을 자동화하고 종이 의존도를 줄이며 중요한 문서에 대한 중앙 집중식 제어 기능을 제공하는 중요한 솔루션으로 두각을 나타냅니다.

전자 학습 관리 시스템(Electronic Learning Management Systems) 소프트웨어를 통한 e-러닝에 대한 선호도가 높아지면서 다양한 산업, 특히 제약 분야에서 큰 관심을 얻었습니다. 유럽 및 기타 경제에서 가장 중요한 부문 중 하나인 제약 산업은 최근 몇 년 동안 급속한 성장을 보였습니다. 팬데믹 이후 의료와 예방접종은 많은 국가에서 최우선 순위가 되었습니다. 그러나 긴급성은 시장에 있는 다른 필수 의약품에도 확대되었습니다. 이렇게 급변하는 환경에서 제약 LMS는 직원이 해당 부문의 증가하는 요구 사항을 처리할 준비가 되어 있는지 확인하기 위해 새로운 직원을 교육하고 인증을 제공하는 데 중요한 기능이 되었습니다. 제약산업이 지속적으로 성장함에 따라 숙련된 인력에 대한 수요도 꾸준히 증가하고 있습니다. 가장 최근의 MarketsandMarkets 보고서에 따르면:

Pharma 4.0이라는 용어는 더 이상 컨퍼런스에서 논의되는 개념이 아닙니다. 그것은 인도에서 현장에서 작동되고 있습니다. 제제 실험실과 제조 부서 전반에 걸쳐 제약 회사는 오래된 시스템을 꾸준히 업그레이드하여 자동화, 데이터 가시성 및 스마트한 인프라를 도입하고 있습니다. 이러한 변화는 생산 지연, 데이터 무결성 실패, 수동 의존성, 규제 조사 증가 등 현지 업계 문제를 해결합니다. 인도 제약 부문이 디지털화와 첨단 기술을 수용함에 따라 몇 가지 필요한 변화가 진행되고 있습니다.






















오늘날 규제가 엄격한 생명 과학 환경에서 조직은 민첩성과 엄격한 규정 준수 요구 사항 사이의 균형을 유지해야 합니다. 로우 코드 QMS 및 MES 솔루션은 광범위한 코딩 없이 신속한 애플리케이션 개발을 지원함으로써 제약 및 생명 공학 회사가 품질, 제조 및 규제 워크플로우를 관리하는 방식을 변화시키고 있습니다. AmpleLogic의 로우 코드 플랫폼은 품질 관리, 제조 실행, 실험실 시스템 및 규제 프로세스를 단일 데이터 기반 프레임워크에 통합하는 통합 에코시스템을 제공합니다. 이를 통해 사일로가 제거되고 실시간 가시성이 향상되며 운영 전반에 걸쳐 더 빠른 의사 결정이 가능해집니다. FDA 21 CFR Part 11, GxP 및 Annex 11과 같은 내장된 규정 준수 프레임워크를 통해 조직은 감사 준비를 보장하는 동시에 배포 일정을 몇 달에서 몇 주로 가속화할 수 있습니다. 플랫폼의 드래그 앤 드롭 구성, 자동화된 워크플로, AI 기반 통찰력은 수동 작업을 줄이고 데이터 무결성을 개선하며 지속적인 개선을 추진하는 데 도움이 됩니다. 로우 코드 QMS 및 MES 솔루션을 채택함으로써 기업은 운영 민첩성을 높이고, 비용을 절감하고, 일관된 규정 준수를 유지할 수 있습니다. 이는 생명 과학 산업의 현대 디지털 혁신을 위한 중요한 전략입니다.

제약 산업은 디지털 혁명의 기로에 서 있습니다. Industry 4.0 기술이 의약품의 제조, 테스트 및 글로벌 유통 방식을 변화시키고 있습니다.

실험실 정보 관리 시스템(LIMS)은 워크플로우 간소화, 컴플라이언스 강화 및 효율성 향상을 위한 현대 제약 실험실의 필수 도구입니다.

제약 세척 검증에서의 규제 컴플라이언스는 협상 불가합니다. 올바른 프로토콜을 이해하고 구현하면 제품 안전과 제조 무결성이 보장됩니다.

제약 제조에서 세척 검증 모범 사례 구현은 제품 품질 유지, 교차 오염 방지 및 GMP 컴플라이언스 보장에 필수적입니다.

올바른 LIMS 소프트웨어 선택은 제약 실험실에 있어 중요한 결정입니다. 올바른 시스템은 실험실 운영을 혁신하고, 컴플라이언스를 강화하며, 결과 도출 시간을 단축할 수 있습니다.

제조 실행 시스템에 인공지능과 머신러닝의 통합이 예측 분석과 실시간 최적화로 제약 생산을 변화시키고 있습니다.

장기 계획은 복잡한 규제 환경, 진화하는 시장 역학 및 기술 변환을 헤쳐나가는 제약 기업들에게 전략적 필수 요소입니다.

aPaaS(서비스형 애플리케이션 플랫폼)와 COTS(상용 기성 솔루션) 및 Low-Code 개발의 융합이 제약 기업들의 애플리케이션 구축 및 배포 방식을 재편하고 있습니다.

제약 및 바이오테크 산업에서 경쟁은 매우 치열하며, 이에 따라 품질 기준이 엄격히 요구됩니다. 이를 위해서는 관련 프로세스에 대한 지속적인 감독과 조사가 필요합니다.

제약 산업 및 기타 다양한 분야에서 환경 조건의 지속적 모니터링은 규제 기준 준수 및 제품 품질 보장을 위해 필수적입니다.

제약 산업은 규제 기대치와 데이터 무결성 및 컴플라이언스 강화의 필요성에 따라 무서류 배치 제조 기록(BMR)으로 점점 더 이동하고 있습니다.

제약 산업의 세척 검증은 규제 기관이 제품 품질 유지 및 교차 오염 방지를 위해 규정한 필수 프로세스입니다.

오늘날 빠르게 변화하는 고도로 규제된 산업에서 제품 검토 소프트웨어는 없어서는 안 될 도구가 되었습니다. 기본 추적 시스템에서 정교한 통합 플랫폼으로 진화했습니다.

운영 탁월성과 품질 보증을 추구하는 과정에서, 확립된 기준에서의 일탈은 예상될 뿐만 아니라 미리 예측됩니다. 성공적인 조직을 진정으로 차별화하는 것은 이러한 일탈을 처리하는 방식입니다.
%20Software.webp&w=3840&q=80)
오늘날 경쟁적인 비즈니스 환경에서 높은 제품 품질을 유지하고 지속적으로 개선하는 것이 성공에 필수적입니다. APR 소프트웨어는 강력한 도구로 부상했습니다.

역동적이고 고도로 규제된 제약 제조 분야에서 첨단 기술의 통합은 핵심 비즈니스 프로세스에서 탁월함을 발휘하고 표준적인 제품 품질을 보장하기 위해 필수적입니다.

연간 제품 품질 검토(APQR)의 핵심 측면에 대한 탐구를 시작하며, 이 조사는 What, Why, Where라는 근본적인 질문들을 깊이 다룹니다.

제약 산업에서 배치 제조 기록(BMR) 검증은 제품 품질, 안전성 및 규제 컴플라이언스를 보장하기 위해 매우 중요합니다. 전통적으로 이 검증은 수동 프로세스였습니다.

빠르게 진화하는 제약 환경에서 자산의 효율적인 관리는 매우 중요합니다. 연구 실험실에서 제조 시설 및 유통 네트워크까지.

제약 산업에서 데이터의 무결성은 매우 중요합니다. ALCOA 원칙—귀속 가능(Attributable), 판독 가능(Legible), 동시 기록(Contemporaneous), 원본(Original), 정확성(Accurate)—은 제조에서 데이터 무결성을 보장하기 위해 확립되었습니다.

학습 관리 시스템(LMS)은 교육, 컴플라이언스 및 전문성 개발을 관리하는 구조화되고 효율적인 방법을 제공하며 제약 산업에서 점점 더 중요해지고 있습니다.
-in-Equipment.webp&w=3840&q=80)
효율성과 정밀성은 생명과학 및 제약 산업의 필수 요소입니다. 이를 통해 기업은 전통적인 수동 프로세스에서 벗어나 비즈니스 프로세스 탁월성을 달성할 수 있습니다.

연간 제품 품질 검토(APQR)는 제약 기업이 제품의 품질 기준을 평가하고, 규제 컴플라이언스를 보장하며, 지속적인 개선을 추진하기 위한 중요한 프로세스입니다.

변경 통제는 제약, 의료기기, 식음료와 같은 규제 산업에서 매우 중요한 프로세스입니다. 제품, 프로세스 및 시스템의 변경을 관리하는 것을 포함합니다.

장비 유지보수는 제약 생산 및 제조 부서의 효율성과 생산성을 보장하는 중요한 측면입니다. 장비 유지보수 소프트웨어가 이 분야를 혁신했습니다.

정밀성, 컴플라이언스 및 효율성이 핵심인 복잡한 제약 제조 세계에서 기업들은 복잡성을 극복하기 위한 혁신적인 솔루션을 모색합니다.

운영 탁월성을 향한 탐구가 전략적 요건이자 지속적인 성공의 초석인 복잡하고 끊임없이 진화하는 현대 산업 환경에서.

제약 산업에서 데이터 무결성 유지는 제품 품질, 환자 안전 및 규제 컴플라이언스를 보장하기 위해 매우 중요합니다. 데이터 무결성은 데이터의 정확성과 일관성을 의미합니다.
-Software-in-Deviation-Handling.webp&w=3840&q=80)
제약 제조 산업에서 효율성, 정확성 및 컴플라이언스는 필수 요소입니다. 표준 운영 절차에서 벗어나는 일탈은 제품 품질을 저하시킬 수 있습니다.

근본 원인 분석(RCA)은 품질 프로세스에서 문제 또는 부적합 사항을 해결하여 부적합의 "근본 원인"을 파악하는 방법입니다. RCA는 원인을 수정하거나 제거하고 예방 조치를 통해 문제의 재발을 방지하는 데 도움이 됩니다.

제약 교육 관리 시스템(TMS)은 GMP 컴플라이언스 교육, 역량 추적 및 규제 감사 준비를 간소화합니다. 현대적인 LMS가 교육 비용을 최대 50% 절감하고, 직원 자격 취득을 가속화하며, 글로벌 운영 전반에서 21 CFR Part 11 컴플라이언스를 보장하는 방법을 알아보세요.

전자 문서 관리 시스템(eDMS)은 제약 및 생명과학 기업의 문서 처리 방식을 근본적으로 변화시켰습니다. FDA 21 CFR Part 11 검증과 명확한 컴플라이언스 이점에도 불구하고, 많은 조직들은 여전히 이 디지털 전환을 완전히 받아들이는 것을 주저합니다.

제약 기업들이 받은 긴 데이터 무결성 경고 목록은 추적 메커니즘 구축 및 강화에 초점을 맞추게 했습니다. 해결책 중 하나는 디지털화입니다. 그러나 현재 과제는 디지털 전환이 예상보다 느리게 진행되고 있다는 것입니다.
제약 제조, 컴플라이언스 및 디지털 전환의 최신 트렌드를 확인하세요.
필요한 곳에 우리가 있습니다. 글로벌로 연결하고, 로컬로 전달합니다.
Melange Tower, 2nd Floor, Wing-C, Patrika Nagar, HITEC City, Madhapur, Hyderabad - 500081, Telangana, India
Block 1, Blanchardstown Corporate Park Ballycoolen Road, Dublin, D15 AKK1
최신 제품 업데이트, 규정 준수 뉴스, 업계 인사이트를 받은 편지함으로 직접 받아보세요.