

CPV3 min read
製薬業界のプロセスパフォーマンス監視に、よりスマートな CPV アプローチが必要な理由
プロセス パフォーマンスを監視する製薬チームは、監査の準備を整えるために、強力な継続的プロセス検証から始めます。 AmpleLogic CPV を使用すると、これがどのように簡単になるかを説明します。
2026-07-31続きを読む
製薬製造、コンプライアンス、AI自動化、ライフサイエンスにおけるデジタルトランスフォーメーションに関する専門家の見解。
プロセス パフォーマンスを監視する製薬チームは、監査の準備を整えるために、強力な継続的プロセス検証から始めます。 AmpleLogic CPV を使用すると、これがどのように簡単になるかを説明します。

ほとんどの製薬会社は、研究、品質、製造、規制機能にわたって 5 ~ 10 の非接続システムを運用しています。 単一の接続されたプラットフォームが、製品ライフサイクル全体でできることをどのように変えるかを説明します。

インド薬局方の第 10 版には、121 の新しいモノグラフと、従来のラボ システムでは追跡するように構築されていなかった段階的な改訂スケジュールが追加されています。 ここでは、QC 研究所にとって IP 2026 が何を意味するのか、そしてデジタル品質システムがこれに対応するために不可欠になりつつある理由を説明します。

手動アクセス制御は、LIMS、eQMS、eBMR、およびその他の検証済みシステム全体には拡張できません。 GAMP 準拠のロールベースのユーザー アクセス管理システムが、検査官が発見する前にコンプライアンスのギャップを埋めるために不可欠になっている理由は次のとおりです。

電子実験室ノートと実験室情報管理システムは異なる目的を果たしますが、これらを組み合わせることで、接続された実験室環境が構築されます。 ELN ソフトウェアと LIMS ソフトウェアを統合することで、製薬およびライフ サイエンスの研究室における効率が向上し、データの整合性が強化され、規制遵守がどのようにサポートされるかをご覧ください。

断片化されたスプレッドシートや従来のツールでは、世界的な製薬規制の要求に対応できません。 提出、監視、コンプライアンスの追跡をカバーする、接続された規制情報管理システムが、あれば便利なものではなく、不可欠なインフラストラクチャになっている理由はここにあります。

最新のプロセス検証ソフトウェアは、製薬メーカーが手動検証を自動化された準拠ワークフローに置き換えるのに役立ちます。 AmpleLogic CVS がどのようにトレーサビリティを向上させ、承認を迅速化し、検証リスクを軽減しながら検査の準備を確保するかを学びましょう。 デジタルプロセス検証が GMP 準拠の製造に不可欠になっている理由をご覧ください。

製薬会社やバイオテクノロジー研究所は年々、より迅速な対応を求められていますが、そのプレッシャーにはコンプライアンスを緩める許可が伴いません。 多くのチームが依然として紙のノートや、10 年間パッチを当ててきたレガシー ソフトウェアに頼っているのであれば、これは困難です。 次世代の電子実験ノートは、ここでの数学を変えます。 これは単なるデジタルラボノートではなくなり、科学組織の接続層として機能し始め、研究、品質、製造、規制の作業を、相互に連携しない一連の個別のステップではなく、1 つのライフサイクルに結び付けます。

一貫した製品品質を維持するには、プロセスを継続的に可視化することから始まります。 AmpleLogic の製薬プロセス監視ソフトウェアが、メーカーが重要なプロセスをリアルタイムで監視し、変動を削減し、コンプライアンスを強化し、継続的プロセス検証 (CPV) を通じて業務効率を向上させるのにどのように役立つかをご覧ください。

製薬工場は依然として、紙ベースの追跡、散在する記録、事後対応型メンテナンスにより時間を浪費し、監査の信頼性を失っています。 キャリブレーションの欠落、システムの切断、機器の健全性のリアルタイムの可視性の欠如により、バッチが失敗するか監査人が質問を開始した後に初めて問題が表面化します。 AmpleLogic の CAPS は、校正とメンテナンスのスケジューリングを自動化し、GMP 準拠の 21 CFR Part 11 に準拠した監査証跡を維持し、eQMS、LIMS、eBMR/MES、DMS、LMS、および SAP/ERP と統合することでこの問題を解決し、検証された機器のみが本番環境に導入されるようにします。 その結果、緊急修理が減り、ダウンタイムが減り、監査の準備がスクランブルではなくレポートのプルになります。

手動のスプレッドシートでは世界的な製薬規制に対応できません。 最新の規制情報管理システムが、提出、追跡、コンプライアンス監視を 1 つのシステムに集中化する方法を次に示します。

効率的な検査室運営は効果的な計画から始まります。 AmpleLogic のデジタル計画ソリューションを使用して、品質管理計画ソフトウェアが製薬会社の QC スケジュールの合理化、検査室リソースの最適化、検査準備の向上にどのように役立つかをご覧ください。

文書化は医薬品製造の根幹です。 すべてのバッチ、途中のすべてのステップ、すべての逸脱 - すべては、どの国から検査しているかに関係なく、規制当局が満足する方法で記録、チェック、ファイルされる必要があります。 それは何年もの間、紙のバッチ記録の山が製造現場から QA まで往復し、再び戻ってきて、そしておそらくもう一度戻ってくることを意味していました。 うまくいきました。 かろうじて。 そしてある時点で亀裂が広がりすぎて、つぎはぎし続けることができなくなりました。 まさにそこに eBMR ソフトウェアが登場します。電子バッチ製造記録システムは、オプションのアップグレードから、工場にとって欠かせないものへと静かに移行しました。

ほとんどの製薬研究室に足を踏み入れると、数十万円もする新品の機器の隣に紙の日誌が置かれているのを目にすることがあるでしょう。 考えてみると不思議です。 研究室は自動化と分析に資金をつぎ込んでいるが、基本的なログブック、つまり誰がいつ何をしたかを記録するものは、依然として、ペンが紐で結ばれた単なるスパイラルノートであることが多い。

品質チームが監査前に SOP の「最終最終バージョン」を見つけ出すために奔走するのを見たことがある人なら、なぜ文書管理がコンプライアンス リーダーを夜も眠れなくさせるのかをすでに知っているでしょう。 AmpleLogic のドキュメント管理システム ソフトウェアは、あらゆるバージョン、承認、署名が検査に耐える必要がある、規制されたライフ サイエンス環境向けに特別に構築されています。 これにより、分散したフォルダーと手動ルーティングが、GMP 文書が実際にどのように移動するかに合わせて設計された GAMP 準拠の監査対応プラットフォームに置き換えられます。

QA プロフェッショナルに毎年最も恐れていることを尋ねると、すぐに「APQR シーズン」が到来します。 ほぼ毎回です。 理論的には、年次製品品質レビューは複雑ではないようです。 1 年間の製造、ラボ、品質データを収集します。 それを分析してください。 製品が期待どおりに機能していることを示します。 十分シンプルですよね?

洗浄プロセス検証ソフトウェアが製薬メーカーの検証の合理化、コンプライアンスのリスクの軽減、業務効率の向上にどのように役立つかをご覧ください。 デジタルプロセス検証システムの導入が、現代の製造業におけるデータの整合性、監査の準備、法規制順守を維持するために不可欠になっている理由を学びましょう。

ほぼすべての製薬工場のエンジニアリング担当者または QA 担当者に校正を持ちかけて、何が起こるかを観察してください。 一時停止があります。 ため息かもしれない。 そして、どんな答えが出ても、大抵は「そうですね、技術的には…」で始まります。これが重要であることは誰もが知っており、誰もそれを異論はしません。 しかし、ほとんどの施設が実際にどのように対応しているかを調べてみると、何年も前から同じ設定が使われていることがわかります。2014 年に誰かが作成したスプレッドシート、バインダーに詰め込まれた紙のログ、チームの半分が無視しているカレンダーのリマインダーなどです。

継続的なプロセス検証は、製薬メーカーが最初の検証を超えてプロセス管理を維持するのに役立ちます。 CPV ソフトウェアがどのように継続的な監視、統計分析、プロセス傾向の早期特定を可能にするかをご覧ください。

バッチリリースは以前はこれほど大したことではありませんでした。 ほとんどの場合、事務手続きフォームが手作業で記入され、部門から部門へと渡され、各拠点で承認され、すべてが正常に完了するとバッチが出荷されることを意味していました。 工場の生産量が少なく、検査の頻度も少なかった頃は、それでも問題ありませんでした。 最近はそれが耐えられません。 検査官は、組み立てに1週間かかるバインダーではなく、その場で確認できる記録を期待して現れます。 患者は、すでに承認された医薬品を必要以上に長く待たされています。 そして、多くの製造業者は現在、複数のサイトを同時に運営していますが、これを紙のフォームや希望で追跡することは基本的に不可能です。 したがって、バイオ医薬品のリーダーの間での本当の会話は、もはや「バッチリリースを最新化すべきか」ということではありません。 コンプライアンス面で誤って何かを壊すことなく、重要な問題となるまで迅速に実行することが重要です。

最新の文書管理ソフトウェアが製薬会社のバージョン管理の問題、承認の遅れ、監査準備の課題をどのように克服するのに役立つかをご覧ください。 一元化された文書管理システムが、規制対象の文書ワークフローに優れた制御、追跡可能性、および効率をもたらす方法を学びましょう。

今日、ほとんどすべての製薬研究室に足を踏み入れると、今でも紙のノート、印刷されたプロトコル、計器パネルに貼り付けられた付箋の山が見つかります。 正確で機密性の高いデータに基づいて運営されるべき業界でこれを見るのは奇妙なことです。 科学者は結果を手書きし、後で別のシステムに同じ数値を再入力します。 そのプロセスのどこかで間違いが紛れ込み、ページが失われます。 そして、監査人が 3 年前に実施した実験の記録を要求すると、誰かが保管箱を漁りながら午後中を費やすことになります。

ほとんどの研究室はすでに LIMS を導入していますが、本当の問題は、それがまだ維持されているかどうかです。 ここでは、QC ラボにおける安定性予測、在庫、STP の処理が AI によってどのように変化しているかを説明します。

品質管理は事後対応型から予測型へと移行しています。 ここでは、AI が eQMS をどのように変えるのか、そして次の目的のために構築されたプラットフォームを選択する際に何を優先すべきかを説明します。

手動による QC 計画は、多くの場合、スケジュールの競合、バッチリリースの遅延、ラボの効率の低下につながります。 最新の **QC 管理システム** が製薬研究所の計画の合理化、リソースの最適化、インテリジェントな QC スケジューリングへのコンプライアンスの維持にどのように役立つかをご覧ください。

APQR シーズン中にほとんどの製薬品質部門に足を運べば、同じことが行われているのを目にするでしょう。 誰かが 3 つのスプレッドシートを開いて、近くにバッチ レコードのスタックを置き、手動で入力するのを待っているレポート テンプレートを持っています。 年次製品品質レビューは、構造化されたデータ主導のプロセスであると考えられています。 実際には、多くのチームにとって、依然として手作業での作業が毎年数週間を費やしています。 データ量が増大し、規制当局の期待が高まるにつれ、その古いやり方はもはや通用しなくなりました。

製薬工場に足を踏み入れると、同じ静かな真実に気づくでしょう。すべては、例外なく、常にその動作を正確に実行する機器に依存しています。 範囲から少し外れているスケール。 予定通りにチェックする人が誰もいなかったセンサー。 メンテナンスログが引き出しのどこかに詰め込まれており、半分埋まっていて期限切れになっています。 これらのいずれかがバッチ全体を危険にさらす可能性があります。 それが校正と予防保守です。表面的にはエンジニアリングの雑務のように見えますが、その下では実際にはコンプライアンス作業です。 一つでも間違えると、機械が故障する危険があるだけでなく、規制上の指摘を受ける危険もあります。

最新の文書管理システム (DMS) が製薬会社の文書管理の合理化、規制遵守の維持、検査への備えの維持にどのように役立つかをご覧ください。 準拠した医薬品文書管理ソリューションに求められる主な機能について学びます。

医薬品製造は、一部の分野では急速に進んでいますが、他の分野では停滞しています。 バッチ記録はデジタルです。 実験室の機器は LIMS と通信します。 しかし、多くの作業現場では、クリップボードがタンクや HVAC パネルの横に吊り下げられ、誰かが測定値を手書きで書き留めるのを待っています。 ハイテク システムとローテク ログブックの間のギャップは、ほとんどの監査の問題の始まりであり、AmpleLogic の電子ログブック (eLogbook) ソフトウェアが変化を生もうとしている点でもあります。

今日、ほとんどの製薬施設に入ると、依然として紙のバッチ記録がファイルキャビネットに詰め込まれているのを目にするでしょう。 署名、スタンプ、レビュー、保管。 そして、その連鎖のどこかで何かがうまくいかなくなります。 署名漏れ。 間違った入力です。 監査の前に消えるページ。 これも例外ではありません。 それは施設のケアが認める以上に起こります。

最新の文書管理システム ソフトウェアが製薬会社のコンプライアンス向上、文書管理の合理化、監査準備の維持にどのように役立つかをご覧ください。 ライフ サイエンス業界の品質、効率、規制上の成功にデジタル ドキュメント管理が不可欠になっている理由を学びましょう。

AI は、eQMS プラットフォームを事後対応型のファイリング システムからプロアクティブな品質パートナーに変えています。 ここでは、何が変化しているのか、何を最初に自動化すべきなのか、そして実際の AI を活用したプラットフォームと流行語との違いは何なのかを説明します。

MACO 計算、リスク評価、検証ワークフローを自動化する AI を活用した洗浄検証ソフトウェアで医薬品コンプライアンスを合理化します。 一元化されたデジタル検証プラットフォームを通じて、監査の準備を改善し、手動の労力を削減し、法規制へのコンプライアンスを確保します。

年次製品品質レビューは規制要件ですが、ほとんどの製薬品質チームにとって、それはひそかに、カレンダー上で最も骨の折れる繰り返しのタスクの 1 つとなっています。 このプロセスは本質的に手動です。 データは切断されたシステム全体に存在します。バッチ記録は 1 か所に、苦情は別の場所に、安定性データは別の場所にあります。 誰かがそれをすべてまとめ、調整し、フォーマットし、一貫したレポートに変換する作業を、製品ごとに毎年繰り返し行う必要があります。 机上では、これはコンプライアンスの実践です。 実際には、何百時間ものスタッフの時間を他の場所に費やせる可能性があります。

製薬会社の古い戦略のスプレッドシート、サイロ化されたデータ、手動分析は、現代のデータ量の重みで壊れつつあります。 この版では、AI が創薬、臨床試験、運営、市場戦略にわたってインテリジェンスをどのように再配線しているのか、そしてなぜ次に勝つ企業は最も多くのデータを持っている企業ではなく、それを最も早く意思決定に反映させている企業であるのかを探ります。

継続的プロセス検証 (CPV) は、製薬メーカーがプロセス管理を維持し、コンプライアンスを確保し、生産に影響を与える前に品質リスクを検出するのに役立ちます。 最新の CPV ソフトウェアがリアルタイム監視、自動化された SPC チャート作成、インテリジェントなアラート、APQR 自動化を可能にし、プロアクティブな品質管理を実現する方法を学びましょう。

製薬業界における安定性試験の手動管理は、プル日の逃し、データのギャップ、コンプライアンスのリスクにつながります。 最新の安定性試験ソフトウェアは、スケジューリングを自動化し、トレーサビリティを向上させ、規制対応の安定性プログラムを保証します。

インテリジェントな QC プランニング ソフトウェアが製薬およびバイオテクノロジー研究所でどのようにスケジューリングの自動化、リソースの最適化、コンプライアンスの向上、所要時間の短縮に役立つかをご覧ください。 よりスマートな計画とキャンペーンベースの実行により、最新の QC 業務がどのように変革され、効率が向上し、バッチ リリースが迅速化されるかを学びましょう。

すべての製薬監査にはストーリーがあります。 FDA 483 の観察や MHRA の調査結果の途中で、通常、捜査官が「誰が、いつ、このシステムにアクセスしたのか?」という一見単純に聞こえる質問をする瞬間があります。 あまりにも多くのライフ サイエンス組織にとって、その質問に答えるには何日もかかるか、まったく明確に答えることができません。 ここでは、内訳と、専用の GxP アクセス管理が実際にどのようなものかを示します。

ソフトウェアを期限内に出荷することは一か八かの賭けです。 信頼性が高く、準拠しており、定義された品質基準に従って構築されたソフトウェアを出荷することで、組織は真の差別化を図ることができます。 ソフトウェア品質管理システム (SQMS) は、品質の計画と保証から継続的な改善と規制監査の準備に至るまで、開発ライフサイクル全体にわたってそれを実現するための構造化されたフレームワークを提供します。

手動によるデータ入力は依然として現代の研究室における最大の課題の 1 つであり、転記エラー、コンプライアンス リスク、業務の非効率につながります。 この記事では、LIMS に機器を統合することで、どのようにデータ収集を自動化し、精度を向上させ、規制遵守をサポートし、製薬、ライフサイエンス、食品検査研究所全体の生産性を向上させるのかについて説明します。

製薬、バイオテクノロジー、医療機器業界におけるドキュメントの管理には、厳格なコンプライアンス、セキュリティ、効率性が必要です。 AmpleLogic ドキュメント管理システムがどのようにドキュメント管理を合理化し、ワークフローを自動化し、安全なデジタル プラットフォームを通じて規制順守を確保するかをご覧ください。


安定性研究は、医薬品がライフサイクル全体にわたって安全で効果的であり、コンプライアンスを遵守していることを保証するために不可欠です。 ただし、これらの研究をスプレッドシートや手動プロセスで管理すると、コンプライアンスのリスク、データの整合性の問題、運用の非効率が生じる可能性があります。 このブログでは、エンドツーエンドの安定性管理の重要性、最新のデジタル安定性プログラムの主要コンポーネント、規制当局の期待、および AmpleLogic の安定性管理プラットフォームのような集中化された準拠ソリューションを使用して製薬企業が安定性研究を合理化する方法について探ります。

AI を活用した電子日誌は、デジタル記録管理とインテリジェントな自動化を組み合わせることにより、医薬品文書を変革しています。 データの整合性や規制遵守の向上から、予測的な洞察やリアルタイムの異常検出の実現に至るまで、AI 主導の eLogbook は、製薬企業が業務を合理化し、手作業を削減し、Pharma 4.0 の取り組みを加速するのに役立ちます。
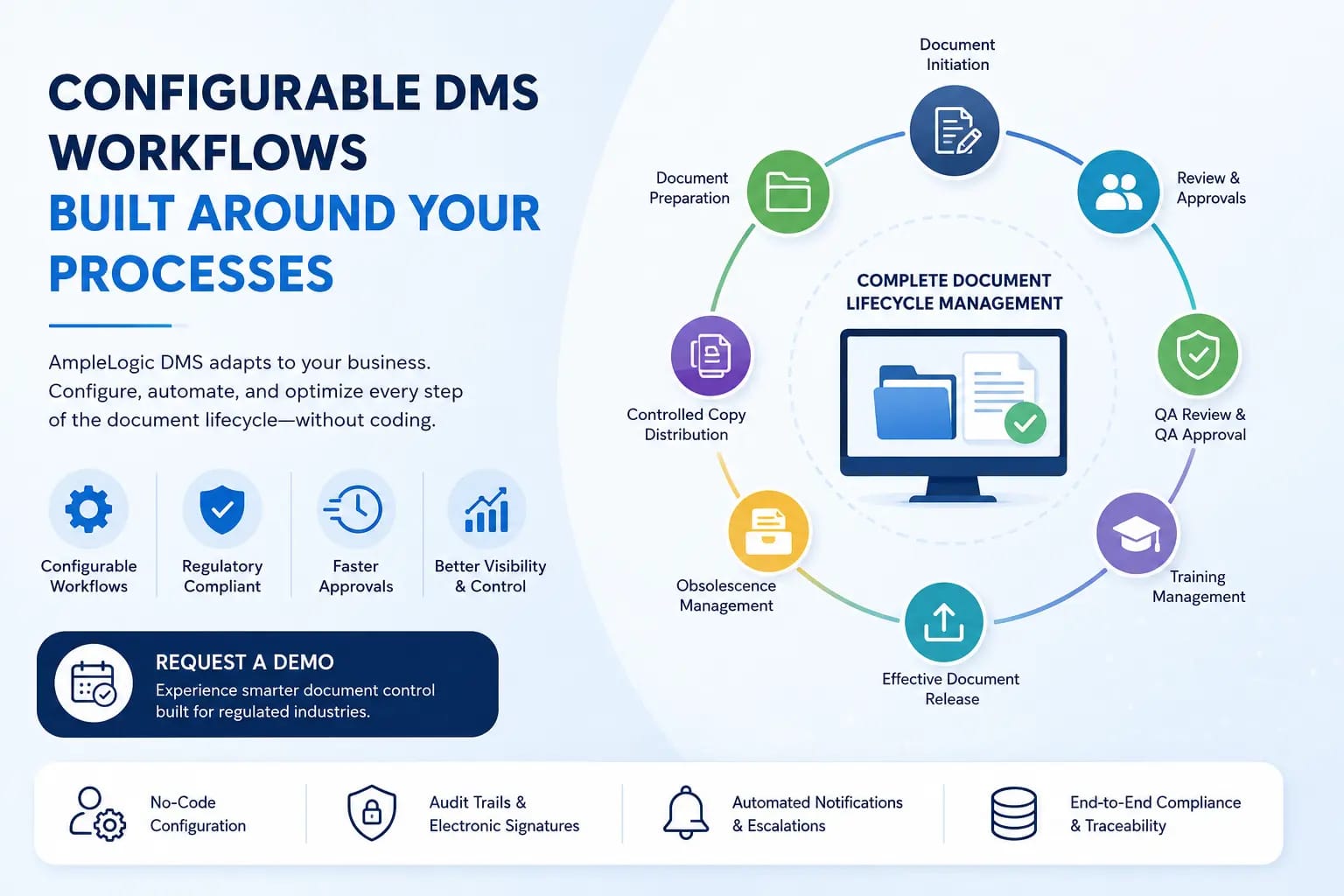
ドキュメントの作成とレビューからトレーニング、定期的なレビュー、陳腐化管理に至るまで、効率的なワークフローの自動化はコンプライアンスと生産性にとって不可欠です。 AmpleLogic DMS が規制業界向けに設計された完全にカスタマイズ可能なワークフローにより、承認のボトルネックをどのように解消するかをご覧ください。

品質はもはや単なる部門ではなく、戦略です。 製薬、製造、ライフ サイエンス全体で規制上の期待が厳しくなる中、インテリジェントな品質管理システム ソフトウェアに投資する組織は、単にコンプライアンスを遵守するだけではありません。 彼らはより速く、よりスマートに、そしてはるかに高い自信を持って動作します。 このガイドでは、最新の QMS プラットフォームの機能、優れた実装と優れた実装の違い、業界と規模に適したソリューションを選択する方法を詳しく説明します。

DMS ソフトウェアにおける一般的なバージョン管理の課題を学び、ドキュメントの正確性、コンプライアンス、コラボレーション、監査の準備を確保するための効果的なソリューションを見つけてください。

製薬工場における予防保守と予知保守の違いと、校正および保守ソフトウェアがどのようにコンプライアンス、機器の信頼性、運用効率を向上させるかを学びます。

プロセスの標準化やシステム統合からコンプライアンスやユーザーの導入に至るまで、製薬企業が eBMR 導入中に直面する最大の課題を探ります。 最新の eBMR ソフトウェアがバッチ製造の合理化、データの整合性の向上、製薬業務のデジタル変革の加速にどのように役立つかを学びましょう。

紙のログブックから電子システムに切り替えることにより、GMP 施設はコンプライアンスを向上し、文書化エラーを減らし、業務効率を向上させることができます。 最新の医薬品製造において、電子ログブックがデータの整合性、監査の迅速化、バッチリリースプロセスの改善、測定可能な ROI をどのようにサポートしているかをご覧ください。

最新の LIMS ソリューションは、自動化、リアルタイムの可視性、データの整合性の向上を通じて、製薬研究所が OOS および OOT 調査を合理化するのに役立ちます。 デジタル調査ワークフローがどのようにコンプライアンスを強化し、根本原因分析を加速し、全体的な品質管理を強化するかをご覧ください。

AI コパイロットが、よりスマートなコンプライアンス、迅速な調査、予測的洞察、品質業務全体にわたるインテリジェントな自動化を可能にして、医薬品の品質管理をどのように変革しているかをご覧ください。 AI を活用した製薬ソフトウェアが、組織の効率向上、手作業の削減、将来に対応したデジタル品質システムの構築にどのように役立つかを学びましょう。

AI を活用した洗浄検証ソフトウェアが、製薬メーカーによる MACO 計算の自動化、GMP コンプライアンスの向上、および手動検証の非効率性の排除にどのように役立っているかをご覧ください。 デジタル検証ワークフローが医薬品製造全体でデータの整合性、監査の準備、業務効率をどのように強化するかを学びます。

革新的なアイデア、イノベーション戦略、テクノロジーとビジネスの未来を形作る革新的なソリューションを評価した、AL アイデアソン 2024 の先見の明のある審査員をご紹介します。



Application Platform as a Service (aPaaS) は、基盤となるインフラストラクチャを処理せずにアプリケーションを構築、デプロイ、管理するための完全な環境を提供するクラウド コンピューティング モデルです。 これにより、企業は開発を加速し、コストを削減し、アプリケーションを効率的に拡張できるようになります。 aPaaS は、ツール、フレームワーク、自動化を提供することで、設計から導入に至るアプリケーションのライフサイクル全体を簡素化し、組織がより迅速にイノベーションを起こし、デジタルファーストの世界で競争力を維持できるように支援します。

GAMP 5 は、製薬業界でコンピュータ化されたシステムを検証するための構造化されたアプローチを提供し、システムが意図された用途に適合し、規制の期待に沿っていることを確認します。 これは、要件からテストまでのライフサイクルベースの検証プロセスをサポートする V-Model などのフレームワークと連携して動作します。 21 CFR Part 11 や EU Annex 11 などの規制では、電子記録と署名に関する厳格なルールが定義されており、システム全体でのデータの整合性、セキュリティ、トレーサビリティが確保されています。 これらの標準を組み合わせることで、組織が検証済みのソフトウェア システムを効率的に開発、実装、保守できる強力なコンプライアンス エコシステムが構築されます。 リスクベースの検証、ライフサイクル管理、規制調整を組み合わせることで、企業はコンプライアンスのリスクを軽減し、製品の品質を向上させ、製薬業務のデジタル変革を加速できます。

ローコード プラットフォームにより、組織はビジュアル インターフェイスと事前構築されたコンポーネントを使用して、最小限の手動コーディングでアプリケーションを設計、開発、展開できます。 製薬会社にとって、規制基準とデータの整合性を維持しながら、プロセスをデジタル化し、ワークフローを自動化し、運用を拡大するための、より迅速でコンプライアンスに準拠した方法を提供します。

バッチ レコード (BMR) 発行およびバッチ番号生成ソフトウェアは、製薬会社が重要な製造プロセスを自動化および制御するのに役立ちます。 安全で追跡可能な効率的なデジタル ワークフローを提供することで、正確なバッチ追跡を保証し、手動エラーを削減し、規制基準への準拠を維持します。

医薬品 QA/QC バッチ プロセスの自動化により、企業は USFDA、MHRA、cGMP 規格への準拠を確保しながら、品質保証と管理活動を合理化できます。 ワークフローをデジタル化し、重要なプロセスを自動化することで、組織はデータの整合性を強化し、手動エラーを減らし、より迅速なバッチリリースを実現できます。

ローコード アプリケーション開発プラットフォームにより、組織はビジュアル ツールと最小限のコーディングを使用して、アプリケーションを迅速に設計、構築、展開できます。 製薬業界では、データの整合性と規制基準を維持しながら、イノベーションの迅速化、コンプライアンスの向上、効率的なワークフローの自動化をサポートします。

ローコードの aPaaS (Application Platform as a Service) プラットフォームは、最小限のコーディングでアプリケーションを迅速に構築、展開、管理するためのクラウドベースの環境を提供します。 これにより、製薬企業はデジタル変革を加速しながら、ワークフローを合理化し、コンプライアンスを確保し、業務を効率的に拡張できるようになります。

GMP SOP トレーニング プランナー、管理、および追跡ソフトウェアは、製薬会社が法規制順守を確保しながら従業員のトレーニング プログラムを効率的に管理するのに役立ちます。 トレーニング スケジュールを自動化し、進捗状況を追跡し、監査に備えた記録を維持することにより、組織は従業員の能力を向上させ、コンプライアンス プロセスを合理化できます。

CAPA 管理ソフトウェアは、製薬企業が是正措置と予防措置を効率的に管理して逸脱に対処し、品質プロセスを改善するのに役立ちます。 ワークフローを自動化し、トレーサビリティを確保することで、コンプライアンスを強化し、リスクを軽減し、継続的な改善をサポートします。

変更管理の自動化および追跡ソフトウェアは、製薬企業が法規制順守を確保しながら、変更を効率的に管理、文書化、追跡するのに役立ちます。 ワークフローを自動化し、監査証跡を維持することにより、トレーサビリティが向上し、リスクが軽減され、品質管理プロセスが強化されます。

QMS 自動化ソフトウェアは、製薬企業やバイオテクノロジー企業が品質プロセスを合理化し、コンプライアンスを管理し、業務効率を向上させるのに役立ちます。 CAPA、逸脱、監査、文書管理などのワークフローを自動化することで、一貫した品質と規制遵守を保証します。

Exchange Server 管理ツールは、組織が電子メール インフラストラクチャを効率的に管理、監視、保護するのに役立ちます。 管理タスクの自動化、パフォーマンスの追跡、セキュリティの強化により、信頼性の高い通信と合理化された IT 運用が保証されます。

ノーコード アプリケーション開発プラットフォームを使用すると、ユーザーはビジュアル インターフェイスと事前構築済みコンポーネントを使用して、コードを記述せずにアプリケーションを構築およびデプロイできます。 製薬業界では、より迅速なイノベーション、コンプライアンスの向上、効率的なワークフローの自動化が可能になります。

Excel の自動化では、ツール、マクロ、ワークフローを使用して反復的なタスクを簡素化し、効率と精度を向上させます。 製薬環境やエンタープライズ環境では、より高速なデータ処理が可能になり、手動エラーが減少し、合理化された運用を通じて生産性が向上します。

環境監視システム (EMS) は、温度、湿度、圧力、微生物汚染などの重要な環境条件を監視する製薬およびバイオテクノロジー施設にとって不可欠です。 リアルタイムのデータ、自動アラート、製造環境全体にわたる完全なトレーサビリティを提供することで、製品の品質、法規制へのコンプライアンス、リスク軽減を保証します。

Quality Suite は、製薬企業やバイオテクノロジー企業が CAPA、逸脱、監査、文書管理などの品質プロセスを管理するのに役立つ統合プラットフォームです。 ワークフローを自動化し、コンプライアンスを確保することにより、効率、データの整合性、および全体的な製品品質が向上します。

資産管理システムは、組織が調達から保守、廃棄に至る資産のライフサイクルを追跡、管理、最適化するのに役立ちます。 製薬環境やエンタープライズ環境では、運用効率を確保し、ダウンタイムを削減し、法規制へのコンプライアンスをサポートします。

プロセス検証ソフトウェアは、製薬会社が製造プロセスで一貫して高品質の製品を生産していることを確認するのに役立ちます。 検証ワークフローを自動化し、詳細な文書を維持することにより、コンプライアンスを強化し、データの整合性を向上させ、効率的なライフサイクル管理をサポートします。

APQR プロセスの遅延、手動のスプレッドシート、またはトレーサビリティの欠如に悩まされていませんか? これらは、年次製品品質レビューがコンプライアンスの準備ができていない可能性がある明らかな兆候です。 上位 5 つの警告シグナルを発見し、デジタル APQR システムがどのように効率を向上させ、監査の準備を整え、製薬業務における法規制順守を強化できるかを学びましょう。

APQR プロセスの非効率性に悩んでいませんか? 不完全なデータ、標準化の欠如、不十分なトレーサビリティなどのよくある間違いは、コンプライアンスや意思決定に影響を与える可能性があります。 これらの落とし穴を回避し、構造化されたデジタル アプローチで年次製品品質レビューを改善する方法を学びましょう。

製薬業界で監査の準備を整えるのに苦労していますか? 手動の APQR プロセスは、多くの場合、遅延、トレーサビリティの低下、コンプライアンスのリスクにつながります。 デジタル APQR システムが、一元化されたデータ、リアルタイムの可視性、自動化された文書化、完全な監査証跡によって監査の準備をどのように向上させるかをご覧ください。


















































FDA ステージ 3 継続プロセス検証 (CPV) は、商業生産中に製造プロセスが検証済みの状態に維持されることを保証する、プロセス検証ライフサイクルの重要なフェーズです。 これは、統計ツールとリアルタイム データ分析を使用して傾向、変動性、潜在的な逸脱を検出することにより、重要なプロセス パラメーター (CPP) と重要な品質属性 (CQA) を継続的に監視することに重点を置いています。 FDA は、ライフサイクル アプローチを採用することで、検証が 1 回限りの活動ではなく、データとリスク管理によって推進される継続的なプロセスであることを強調しています。 CPV は、プロセス パフォーマンス データを年次製品レビューと管理上の意思決定に統合し、プロアクティブな品質保証と規制遵守を可能にします。 AmpleLogic のような高度なデジタル ソリューションを使用すると、組織は CPV の実装を合理化し、データの整合性を強化し、進化する FDA の期待への継続的なコンプライアンスを確保できます。


21 CFR Part 11 に準拠した文書管理システムは、デジタル業務に移行する製薬およびライフ サイエンス組織にとって不可欠です。 21 CFR Part 11 は、電子記録と電子署名が信頼できるものであり、紙ベースの文書と同等であるとみなされる基準を確立しています。 準拠した文書管理システムを実装することにより、組織は完全な監査証跡、アクセス制御、検証済みワークフローを維持しながら、規制対象文書を安全に作成、保存、取得、管理できます。 これらのシステムは、追跡可能で改ざん防止の記録を提供することで、データの整合性を確保し、不正な変更を防止し、規制検査をサポートします。 AmpleLogic のようなソリューションを使用すると、企業は文書管理プロセスをデジタル化し、電子署名を使用して承認を自動化し、FDA 規制へのシームレスなコンプライアンスを達成できます。これにより、最終的に業務効率が向上し、コンプライアンス リスクが軽減され、品質および規制機能全体にわたる監査の準備が確保されます。

FDA 510(k) 認可プロセスは、米国市場への参入を目指す医療機器メーカーにとって重要な規制経路です。 市販前通知とも呼ばれるこの制度は、安全性と有効性の点で、自社のデバイスがすでに合法的に市販されている述語デバイスと「実質的に同等」であることを企業に証明することを義務付けています。 主にクラス II および一部のクラス I デバイスに適用される 510(k) プロセスには、デバイスの説明、使用目的、ラベル付け、リスク分析、性能テスト データなどの詳細な文書の提出が含まれます。 FDA はこの情報を評価して、デバイスが認可および商業流通に関する規制基準を満たしているかどうかを判断します。 AmpleLogic のようなデジタルで自動化されたコンプライアンス ソリューションを採用することで、メーカーは文書管理を合理化し、データの整合性を確保し、510(k) 提出ライフサイクルを加速することができます。これにより、規制コンプライアンスを維持しながら、遅延を削減し、監査の準備を改善し、革新的な医療機器をより迅速に市場に投入することができます。

ラボの計画とスケジュールをデジタル化することは、増加する作業負荷、複雑なテスト要件、厳格なコンプライアンス基準に直面している現代の QC ラボにとって不可欠です。 スプレッドシート、ホワイトボード、または基本システムに依存することが多い従来のアプローチでは、何千ものテストを効率的に調整する必要がある、ますます複雑になる検査室の運営を管理するのに苦労しています。 デジタル ソリューションを採用することで、研究室は効率を大幅に向上させ、所要時間を短縮し、リソースの利用を最適化できます。 実証済みの利点としては、テストの実行の高速化、在庫要件の削減、優先順位ベースのテスト ワークフローへの遵守の向上などが挙げられます。 主なベスト プラクティスには、シミュレーション用のデジタル ツイン モデリングの実装、クリティカル パス テストの優先順位付け、キャンペーン ベースの実行の活用、リソース プランニングの統合、リアルタイムの適応スケジューリングの有効化などが含まれます。 これらの戦略により、可視性が向上し、ボトルネックが最小限に抑えられ、意思決定が強化されます。 AmpleLogic のようなプラットフォームを使用すると、組織はインテリジェントなスケジューリング、AI 主導の洞察、リアルタイム監視を統合して、ラボの運用を変革し、コンプライアンスの確保、生産性の向上、ライフ サイエンス環境の継続的改善のためのスケーラブルな基盤の構築を実現できます。

バッチリリースは医薬品製造において最も重要かつ高度に規制された段階の 1 つであり、製品の流通前にすべての生産および品質データの徹底的なレビューと承認が必要です。 従来の手動レビュープロセスは時間がかかり、エラーが発生しやすく、追跡が難しいことが多く、遅延やコンプライアンスのリスクにつながります。 AmpleLogic の PQR ソフトウェアは、レビュー プロセス全体を標準化および合理化する自動バッチ リリース チェックリストを導入することで、これらの課題に対処します。 バッチ製造記録 (BMR)、逸脱、CAPA、品質システムからのデータを、構造化された監査対応のワークフローに統合し、すべてのステップでの完全性とトレーサビリティを確保します。 構成可能な SOP ベースのチェックリスト、自動検証フラグ、デジタル署名、リアルタイム ダッシュボードを備えたこのプラットフォームにより、QA チームは問題を早期に検出し、手作業を削減し、バッチ承認を迅速化できます。 これにより、データの整合性が向上し、コンプライアンスが強化され、市場投入までの時間が短縮され、バッチ リリースがより効率的で信頼性が高く、検査に対応できるようになります。

FDA 警告書は、製薬会社が製品の品質と患者の安全を確保するために重要な現行適正製造基準 (CGMP) などの規制基準を遵守していない場合に発行されます。 これらの手紙では、積極的に対処しなければ、製品のリコール、業務の中断、風評被害につながる可能性のあるコンプライアンスのギャップが繰り返し発生することが強調されることがよくあります。 FDA の警告書で特定される一般的な問題には、不適切な原材料検査、不十分なサプライヤー資格、適切な文書化と品質監督の欠如、不十分なプロセスと洗浄の検証、実験室の管理とデータの整合性の失敗などが含まれます。 さらに、安定性試験、バッチ記録管理、環境モニタリング、および変更管理プロセスの不備が規制当局によって頻繁に指摘されています。 これらの課題に対処するために、組織はデジタル品質管理システム、LIMS、およびトレーサビリティを確保し、コンプライアンスを強制し、人的エラーを削減する自動化されたワークフローをますます導入しています。 AmpleLogic が提供するような統合デジタル ソリューションを導入することで、企業はリスクを積極的に特定し、品質プロセスを合理化し、継続的な監査の準備を維持することができ、最終的には費用のかかる FDA 警告書を回避し、長期的な規制遵守を確保できます。

食品および飲料の研究所は、生産ライフサイクル全体にわたって製品の安全性、品質、規制遵守を確保する上で重要な役割を果たしています。 原材料のテストから完成品の検証に至るまで、これらのラボでは、複雑なワークフロー、大量のデータ、および厳しい食品安全基準を管理する必要があります。 食品および飲料ラボ向けの LIMS ソフトウェアは、サンプル追跡を合理化し、データ キャプチャを自動化し、ISO 17025、HACCP、食品安全基準などの世界的な規制への準拠を保証する集中プラットフォームを提供します。 これにより、原材料の調達から最終製品のリリースに至るまでのエンドツーエンドのトレーサビリティが可能になり、手作業によるエラーが減り、データの整合性が向上します。 バッチおよびロットのトレーサビリティ、機器の統合、自動化されたワークフロー、リアルタイムレポートなどの高度な機能を備えた最新の LIMS ソリューションは、研究所の業務効率の向上、試験サイクルの加速、監査の準備の維持を可能にします。 AmpleLogic のようなプラットフォームを導入することで、組織は食品品質管理プロセスを変革し、一貫性があり、準拠した高品質の製品結果を保証できます。

製薬企業やライフサイエンス企業が規制の圧力と運用の複雑さの増大に直面するにつれ、紙ベースの QMS とデジタル QMS の間の議論がこれまで以上に重要になってきています。 従来の紙ベースのシステムは、かつては標準でしたが、現在では手動文書化、承認の遅さ、監査準備の維持の難しさなどの非効率性に悩まされています。 紙ベースの QMS は、文書の置き間違い、バージョン管理の問題、品質プロセスの可視性の制限などのリスクを引き起こすことが多く、規制の厳しい環境ではコンプライアンス管理がより困難になります。 対照的に、デジタル QMS (eQMS) はすべての品質プロセスを一元化し、ワークフローを自動化し、正確で追跡可能なデータへのリアルタイム アクセスを保証します。 デジタル QMS ソリューションには、自動化された文書管理、安全な監査証跡、電子署名、リアルタイム レポート ダッシュボードなどの大きな利点があります。 これらの機能により、FDA 21 CFR Part 11 や GMP 標準などの規制への準拠を確保しながら、データの整合性が強化され、人的エラーが削減され、全体的な運用効率が向上します。 AmpleLogic のようなプラットフォームを使用すると、組織は事後対応的な手動の品質管理から、プロアクティブなデータ駆動型のアプローチに移行でき、より迅速な意思決定、コンプライアンスの向上、拡張可能な品質運用が可能になります。 今日のデジタル時代において、最新の QMS は単なるアップグレードではなく、競争力と優れた規制を維持するために戦略的に必要です。

ライフ サイエンス業界における文書の管理は、厳しい規制要件、大量のデータ、完全なトレーサビリティの必要性により複雑です。 コンプライアンス文書管理システム (DMS) は、組織が文書プロセスをデジタル化、制御、標準化すると同時に、FDA 21 CFR Part 11、EU Annex 11、GxP ガイドラインなどの世界的な規制を確実に順守するのに役立ちます。 従来の紙ベースのシステムや断片化されたシステムは、多くの場合、非効率性、バージョン管理の問題、コンプライアンスのリスクにつながります。 対照的に、最新の電子文書管理システムは、作成とレビューから承認、リリース、アーカイブに至る文書のライフサイクルを一元的に管理し、正確性、一貫性、監査への対応を保証します。 電子署名、監査証跡、自動ワークフロー、AI を利用した検索などの高度な機能を備えた AmpleLogic のようなプラットフォームにより、ライフ サイエンス組織はデータの整合性を向上させ、コラボレーションを強化し、手作業を削減できます。 文書管理プロセスをデジタル化することで、企業はより迅速な承認、コンプライアンスの可視性の向上、高度に規制された環境における優れた規制のためのスケーラブルな基盤を実現できます。

製薬会社は、コンプライアンス、イノベーション、業務効率を維持しながら IT コストを削減するというプレッシャーが増大しています。 増加するインフラストラクチャ費用、レガシー システム、複雑な規制要件により、IT は業界で最も重要なコスト センターの 1 つとなっています。 戦略的な IT コスト削減では、システム効率の向上、冗長性の排除、最新テクノロジーの活用により、経費を削減するだけでなく最適化することに焦点を当てています。 たとえば、IT インフラストラクチャの統合、アプリケーションの合理化、サポート サービスのアウトソーシングにより、運用オーバーヘッドを大幅に削減できます。 調査によると、IT システムとベンダー管理を合理化するだけで、短期間で総 IT コストを約 30% 削減できることがわかっています。 主な戦略には、クラウドベースのプラットフォームの採用、手動プロセスの自動化、品質およびコンプライアンス システムの統合、複数の非接続ツールへの依存の削減などが含まれます。 さらに、特殊な機能をアウトソーシングし、リソースの使用率を改善することで効率が大幅に向上し、これらのアプローチを組み合わせて劇的なコスト削減を達成している組織もあります。 AmpleLogic のようなプラットフォームを使用することで、製薬会社はシステムを統合し、ワークフローを自動化し、データの可視性を向上させることができ、より賢明な意思決定とスケーラブルなデジタル変革を可能にします。 自動化、統合、プロセスの最適化を適切に組み合わせて実装することで、組織は IT コストを大幅に削減しながら、コンプライアンス、俊敏性、全体的なビジネス パフォーマンスを向上させることができます。

製薬業界で一貫した製品品質と規制遵守を確保するには、効果的な年次製品品質レビュー (APQR) を実施することが重要です。 ただし、組織はプロセスが複雑で時間がかかり、エラーが発生しやすくなる複数の課題に直面することがよくあります。 主な課題の 1 つは、LIMS、QMS、ERP、スプレッドシートなどのシステム全体に断片化されたデータが分散しており、データの集計が困難になり、エラーが発生しやすくなることです。 さらに、手動でデータを処理すると、不整合、重複、データの整合性の問題が発生し、APQR レポートの精度が損なわれる可能性があります。 その他の一般的な課題としては、規制遵守のプレッシャー、傾向分析のための限られた分析能力、非効率な部門間のコラボレーション、監査中のトレーサビリティの欠如などが挙げられます。 これらの問題は、多くの場合、遅延、手作業の増加、レビュー プロセスの有効性の低下を引き起こします。 これらの課題を克服するために、製薬会社はデータ ソースを統合し、ワークフローを自動化し、リアルタイム分析を提供するデジタル ソリューションを導入しています。 標準化された SOP を実装し、データ ガバナンスを改善し、部門間のコラボレーションを可能にし、高度な APQR ソフトウェアを活用することで、効率が大幅に向上し、コンプライアンスを確保し、APQR をプロアクティブなデータ主導型の品質プロセスに変えることができます。

医療機器としてのソフトウェア (SaMD) とは、物理的な医療機器の一部ではなく、診断、治療、監視などの医療機能を実行することを目的としたソフトウェアを指します。 国際医療機器規制者フォーラム (IMDRF) や FDA などの世界的な規制機関によると、SaMD はコンピューター、モバイル デバイス、クラウド環境などの汎用プラットフォーム上で動作し、臨床的に関連した結果を提供します。 SaMD ソリューションは、画像診断分析や臨床意思決定支援システムから、患者の状態を監視するモバイル医療アプリケーションに至るまで、ヘルスケア全体で広く使用されています。 これらのシステムは、患者の安全性と有効性を確保するために、リスク分類、検証、サイバーセキュリティ、データ整合性基準などの厳格な規制要件に準拠する必要があります。 デジタルヘルスとAI主導のテクノロジーの急速な成長に伴い、SaMDはより迅速な診断、個別化された治療、リアルタイムモニタリングを可能にすることでヘルスケアの提供方法を変革しています。 ただし、コンプライアンス、品質保証、ライフサイクル管理に関連する課題も生じます。 AmpleLogic のようなプラットフォームは、組織が検証を合理化し、法規制へのコンプライアンスを確保し、医療機器ソフトウェアのライフサイクル全体を管理するのに役立ち、安全性とコンプライアンスの基準を維持しながら、より迅速なイノベーションを可能にします。

医療機器の品質管理システム (QMS) は、設計、開発から製造、市販後調査までのライフサイクル全体を通じて、製品の品質、安全性、規制順守を確保するために設計されたプロセス、手順、および責任の構造化されたフレームワークです。 医療機器メーカーは、ISO 13485、EU MDR、FDA の品質管理システム規制 (QMSR) などの厳格な世界規制に準拠する必要があります。これらの規制は、一貫した製品の品質と患者の安全を確保するための国際基準に準拠しています。 堅牢な QMS には、設計管理、リスク管理、サプライヤー品質管理、CAPA (是正措置および予防措置)、トレーニング管理、監査プロセスなどの重要な要素が統合されています。 これらのコンポーネントは、組織がトレーサビリティを維持し、リスクを最小限に抑え、業務全体にわたる継続的な改善を保証するのに役立ちます。 AmpleLogic のようなデジタル QMS ソリューションを採用することで、医療機器企業は品質プロセスを合理化し、コンプライアンス ワークフローを自動化し、データの整合性を向上させることができます。 これにより、規制の厳しい業界において、より迅速な規制承認、監査対応の強化、安全で高品質の医療機器の一貫した提供が可能になります。

人工知能は、従来の手動プロセスの非効率性に対処することで、製薬およびライフサイエンス業界の品質管理システム (QMS) を急速に変革しています。 従来の QMS は、データの断片化、調査の遅れ、品質イベント全体の可視性の制限に悩まされることが多く、コンプライアンスや運用効率に影響を及ぼします。 AI を QMS に統合することで、組織は CAPA、逸脱管理、変更管理、監査管理などの重要な品質プロセスを自動化できます。 AI を活用したシステムは、機械学習と予測分析を活用して傾向を特定し、異常を検出し、是正措置を推奨します。これにより、根本原因の迅速な分析とプロアクティブなリスク軽減が可能になります。 AI 主導の QMS は、リアルタイムの洞察を提供し、データの精度を向上させ、手動介入を減らすことで意思決定を強化します。 予測的な OOS/OOT 検出、自動化された苦情処理、インテリジェントなリスク評価などの機能は、組織が製品の品質を向上させ、法規制順守を確保し、運用コストを削減するのに役立ちます。 AmpleLogic のようなプラットフォームを使用することで、製薬会社は完全に統合された AI を活用した品質エコシステムを導入でき、世界的な規制への厳格な準拠を維持しながら、ワークフローを合理化し、承認を迅速化し、大幅な効率向上を実現できます。

医療機器の文書管理は、品質と規制遵守の重要な要素であり、設計、製造、品質管理に関連するすべての文書がライフサイクル全体にわたって適切に作成、レビュー、承認、維持されることを保証します。 これは、高度に規制された環境において製品の安全性、トレーサビリティ、監査の準備を維持する上で重要な役割を果たします。 FDA 21 CFR Part 820、ISO 13485、EU MDR などの規制枠組みにより、組織は文書の承認、バージョン管理、配布、および変更管理のための正式な手順を確立する必要があります。 これらの規制により、最新の承認された文書のみが使用され、データの整合性を維持するためにすべての変更が追跡および検証されることが保証されます。 堅牢な文書管理システムにより、組織はデバイス マスター レコード (DMR)、デバイス履歴レコード (DHR)、品質手順などの重要な記録を完全なトレーサビリティで管理できます。 AmpleLogic のようなデジタル ソリューションを導入することで、企業は文書ワークフローを自動化し、コンプライアンスを強化し、安全な監査証跡を維持し、業務効率を向上させることができ、規制検査への備えと長期にわたる優れた品質を確保できます。

品質主要業績評価指標 (KPI) は、医薬品製造における品質管理システム (QMS) の有効性を評価するために使用される重要な指標です。 これらの KPI は、製品の品質、プロセスの効率、規制順守に関する測定可能な洞察を提供し、組織がデータに基づいた意思決定を行い、継続的な改善を確実に行うのに役立ちます。 医薬品製造において一般的に追跡される KPI には、バッチ不合格率、逸脱率、CAPA の有効性、規格外 (OOS) インシデント、調査のサイクル タイムなどが含まれます。 これらの指標は、プロセスの非効率性を特定し、品質の問題を早期に検出し、タイムリーな是正措置を確実に行うのに役立ちます。 このような KPI を監視することは、GMP 基準への準拠を維持し、一貫した製品品質を確保するために重要です。 さらに、RFT (Right First Time)、不良率、プロセス サイクル タイムなどの指標により、製造パフォーマンスと運用効率に関する洞察が得られます。 これらの KPI を分析することで、組織は手戻りを減らし、コストを最小限に抑え、全体的な生産成果を向上させることができます。 AmpleLogic のようなデジタル プラットフォームを使用すると、製薬会社は KPI 追跡を自動化し、リアルタイム ダッシュボードを生成し、予測的な洞察を得ることができ、品質管理をプロアクティブなデータ駆動型の機能に変え、コンプライアンス、効率、製品の信頼性を向上させることができます。

安定性研究は医薬品開発の重要な要素であり、規定された環境条件下で医薬品の有効期間全体にわたってそのアイデンティティ、強度、品質、純度が維持されることを保証します。 これらの調査は、ICH ガイドラインに沿った有効期限、保管要件、および規制遵守を確立するのに役立ちます。 回帰分析は、劣化パターンをモデル化し、製品の保存期間を予測することにより、安定性データを解釈する上で重要な役割を果たします。 回帰技術は、温度、湿度、光への曝露などの要因によって影響を受ける傾向を分析することにより、有効期限の正確な推定を可能にし、医薬品の品質管理におけるデータ主導の意思決定をサポートします。 ANOVA や ANCOVA などの高度な統計手法は、条件間の重大な変動を特定し、外部変数を調整することにより、安定性分析をさらに強化します。 これらのアプローチにより、安定性予測の精度と信頼性が向上し、一貫した製品パフォーマンスと法規制への準拠が保証されます。 AmpleLogic の安定性試験管理ソフトウェアなどのデジタル ソリューションを活用することで、組織はワークフローを自動化し、リアルタイムの傾向を監視し、インテリジェントな統計モデルを適用することができ、安定性試験をより効率的で正確かつ準拠したプロセスに変え、医薬品の長期的な信頼性を保証します。

逸脱管理は医薬品品質システムの重要な要素であり、規制基準に準拠して不適合が適切に特定、調査、解決されることを保証します。 ただし、従来のアプローチは時間がかかり、手動でリソースを大量に消費することが多く、調査の遅れやコンプライアンス リスクの増大につながります。 人工知能は、データ収集、根本原因分析、CAPA 推奨事項などの重要なステップを自動化することにより、逸脱管理を変革しています。 AI を活用したシステムは、過去の逸脱を分析し、プロセスや機器全体のパターンを特定し、質の高いチームがより迅速かつ正確な意思決定を行えるようにするデータ駆動型の洞察を提供します。 AmpleLogic の QMS などの AI 対応ソリューションを使用すると、組織は調査のタイムラインを大幅に短縮し、CAPA の有効性を向上させ、逸脱の再発を最小限に抑えることができます。 これらのシステムはトレーサビリティを強化し、監査の準備を確実にし、事後的な問題処理から予測的でインテリジェンス主導のコンプライアンスへの移行というプロアクティブな品質管理を可能にします。

安定性ソフトウェアは、医薬品開発ライフサイクル全体を通じてコンプライアンスを確保し、製品の品質を維持する上で重要な役割を果たします。 安定性研究は、医薬品がさまざまな環境条件下でそのアイデンティティ、強度、品質、純度をどのように維持するかを判断するために不可欠であり、規制当局の承認とライフサイクル管理の基礎となっています。 最新の安定性管理ソフトウェアを使用すると、組織は ICH 準拠の安定性研究を効率的に設計、実行、監視できます。 これらのシステムは、プロトコル管理、サンプル採取スケジュール、環境モニタリング、統計的傾向分析などの重要なプロセスを自動化し、正確な保存期間予測と堅牢な規制への提出を保証します。 安定性ワークフローをデジタル化することで、製薬会社は手動追跡を排除し、データの整合性を向上させ、FDA 21 CFR Part 11 および GxP 要件に合わせた完全な監査証跡を維持できます。 高度なプラットフォームは LIMS および QMS システムとも統合されており、研究の進行状況をリアルタイムで可視化し、積極的な意思決定を可能にします。 AmpleLogic のようなソリューションを使用すると、組織は安定性研究の管理を合理化し、コンプライアンスを強化し、一貫した製品品質を確保することができ、最終的に開発スケジュールを加速し、高度に規制された環境における全体的な運用効率を向上させることができます。























製薬業界における年次製品品質レビュー (APQR) は、継続的な改善と規制順守を確保するために、製品の品質、製造の一貫性、逸脱、コンプライアンスを長期にわたって評価する GMP 要件です。

製薬業界の製品品質レビュー (APQR) における主な課題には、データ統合の問題、一貫性のない文書化、逸脱管理、法規制遵守のギャップ、および品質、効率、継続的改善に影響を与える限定的な傾向分析などが含まれます。

製薬における手動 APQR と自動 APQR では、効率、データの正確性、コンプライアンス、拡張性における主な違いを調査し、製薬会社のリーダーが品質レビューと継続的改善のための適切なアプローチを選択できるようにします。

AmpleLogic が Gartner の検査情報管理システム (LIMS) マーケット ガイドに掲載されたことは、ライフ サイエンス テクノロジー分野における同社の影響力の増大を裏付けています。 AmpleLogic は、ローコードの AI を活用したプラットフォームにより、研究室がワークフローを合理化し、データの整合性を強化し、デジタル変革の取り組みを加速できるようにします。 この評価は、現代の研究室向けに革新的で準拠したスケーラブルなソリューションを提供するという同社の取り組みを反映しています。

APQR プロセス内のバッチリリースを自動化すると、製薬会社の品質とコンプライアンスの管理方法が変わります。 従来、バッチリリースでは、製品が流通に適していることを確認するために、製造記録と品質記録を手作業で広範にレビューする必要がありました。 年次製品品質レビュー (APQR) では、1 年間にわたるすべてのバッチのデータを統合して、傾向を特定し、一貫した製品品質を確保します。 自動化を APQR に統合することで、組織はバッチ レビュー ワークフローをデジタル化し、チェックリスト主導の検証を実装し、逸脱、CAPA、品質指標のリアルタイム追跡を可能にすることができます。 これにより、データの整合性と監査の準備が向上するだけでなく、リリース サイクル タイムも大幅に短縮されます。 AmpleLogic のようなプラットフォームを使用することで、製薬会社は事後対応の品質チェックからプロアクティブなデータ駆動型のバッチリリース決定に移行し、コンプライアンスを損なうことなくより迅速な承認を確保できます。














分析テストにおける規格外 (OOS) 結果を調査するための包括的なガイド。 AmpleLogic の eQMS を使用した構造化されたワークフロー、根本原因分析、規制準拠のアプローチを学び、規制産業におけるデータの整合性、品質保証、効果的な CAPA 管理を確保します。

AmpleLogic の eQMS が製薬業界の品質管理システムのライフサイクルをどのように簡素化するかをご覧ください。 導入から継続的な改善に至るまで、シームレスなコンプライアンスを達成し、製品の品質を向上させ、高度でスケーラブルなプラットフォームを使用して重要な品質プロセスをデジタル化します。

ライフサイエンスにおける人的エラーは、コンプライアンスのリスクやコストのかかる逸脱につながる可能性があります。 ワークフローを合理化し、データキャプチャを自動化し、製薬およびバイオテクノロジー業務全体の正確性を確保する 6 つの重要なデジタル ツールをご覧ください。

人的エラーは依然としてサイバーセキュリティ侵害の主な原因の 1 つであり、業界全体のインシデントの最大 95% を占めています。 ライフサイエンスでは、たとえ軽微なミスでも機密データが侵害され、コンプライアンスが混乱し、患者の安全に影響を与える可能性があるため、堅牢なデジタル システムが不可欠となっています。

Cipla が AmpleLogic の高度な UAM ソリューションを使用して製薬業務におけるユーザー アクセス管理をどのように合理化したかをご覧ください。 このケース スタディでは、コンプライアンスの向上、ユーザー プロビジョニングの自動化、GxP 要件に合わせた安全で監査対応のアクセス制御に焦点を当てています。

医薬品製造における MACO 計算と洗浄検証に関する規制要件を理解します。 このガイドでは、FDA、EMA、CDSCO、WHO などの世界的な機関が、コンプライアンス、患者の安全、監査の準備を確保するために、PDE および用量ベースのアプローチを使用して、科学的に正当化されたリスクベースの残留制限をどのように期待しているかについて説明します。

人的ミスは依然として医薬品製造におけるコンプライアンス問題の主な原因の 1 つです。 eBMR、QMS、LIMS などのデジタル ソリューションを導入することで、組織は手動の非効率性を排除し、データの精度を向上させ、自動化、リアルタイム監視、標準化されたワークフローを通じて規制遵守を確保できます。

Teva が AmpleLogic の eQMS を活用して品質プロセスをデジタル化し、コンプライアンスを向上させ、手動作業負荷を軽減した方法をご覧ください。 このケーススタディでは、統合デジタル プラットフォームが製薬業界でどのように業務の高速化、監査への対応力の向上、拡張性の高い品質管理を可能にしたのかを明らかにします。

古いアプリケーション ソフトウェアは、組織を重大なセキュリティ リスク、コンプライアンス違反、運用の非効率にさらす可能性があります。 この記事では、レガシー システムが製薬などの規制業界にどのような影響を与えるか、また、最新の安全なデジタル ソリューションへのアップグレードが不可欠である理由について説明します。 AmpleLogic が企業のコンプライアンス、安全性、将来への対応をどのように支援するかを学びましょう。

AmpleLogic の化粧品業界向け LMS は、トレーニングを合理化し、法規制への準拠を確保し、従業員の能力を強化します。 AI 主導の自動化、SOP 管理、GMP 対応機能により、化粧品会社は高度に規制された環境で品質、安全性、監査の準備を維持できます。

FDA ソフトウェア検証プロセスは、製薬業界のコンピュータ化されたシステムが規制要件を満たしながら一貫して意図したとおりに動作することを保証するために不可欠です。 このブログでは、検証ライフサイクルの段階、文書化、リスクベースのアプローチ、およびデジタル ツールがコンプライアンス、データの整合性、監査の準備状況の達成にどのように役立つかについて説明します。

AmpleLogic の QC プランニングおよびスケジューリング ソフトウェアは、製薬研究所にインテリジェントなプランニング、自動スケジューリング、キャンペーン ベースの実行を提供します。 リソースの利用率が向上し、所要時間が短縮され、リアルタイムの可視性と最適化されたラボのワークフローを通じてコンプライアンスが確保されます。

実用的な eLogbook チェックリストは、QA、QC、および生産チームが文書を標準化し、人的エラーを削減し、GMP コンプライアンスを維持するのに役立ちます。 製薬会社はログブックをデジタル化することで、データの整合性を向上させ、監査の準備を整え、日々の業務ワークフローを合理化することができます。

OCuSOFT が AmpleLogic の eQMS を活用してエンドツーエンドの品質プロセスをデジタル化し、紙ベースの記録を 85% 削減しながら、可視性、コンプライアンス、監査の準備を向上させた方法を学びましょう。 このソリューションにより、規制された環境における文書管理、CAPA、監査のワークフローが合理化されました。

FDA 483 の観察に対応するには、タイムリーで構造化された、証拠に基づいたアプローチが必要です。 組織は、根本原因の分析、是正および予防措置 (CAPA) によってそれぞれの観察結果に対処し、警告書を回避してコンプライアンスを強化するためのスケジュールを明確にする必要があります。

施設の変更から QA を除外すると、コンプライアンスのギャップ、検証の失敗、規制リスクの増加につながる可能性があります。 QA は、影響の評価、適切な文書化の確保、構造化された変更管理およびリスク管理プロセスを通じて GMP 基準を維持する上で重要な役割を果たします。

医薬品バッチ発行手順は、GMP 準拠、トレーサビリティ、製品品質を確保するために重要です。 バッチ製造記録 (BMR) の発行から原材料の検証、すべてのステップの文書化に至るまで、これらのプロセスは各バッチの完全な監査証跡を提供し、規制検査をサポートします。

不完全なバッチ記録は FDA 監査中の重大な危険信号であり、多くの場合 483 件の観察や警告書につながります。 署名の欠落、不明確な入力、データのギャップにより、製品の品質、トレーサビリティ、データの整合性に関する懸念が生じ、コンプライアンスのためには堅牢な文書化の実践が不可欠となります。
















ADC Therapeutics が AmpleLogic を使用して標準管理をデジタル化し、コンプライアンスを向上させ、手作業を削減し、データの可視性と効率を向上させた方法をご覧ください。

製薬業界でよくある MHRA データ整合性障害と、それを防ぐ方法について学びます。 コンプライアンスを向上させ、ALCOA+ の原則を確保し、ベスト プラクティスに従って監査の準備を整えます。

製薬における無菌 LIMS が OSD LIMS とどのように異なるかをご覧ください。 ワークフロー、コンプライアンスのニーズ、データ管理を比較して、適切なソリューションを選択します。

人的エラーが製薬業界のデータ整合性にどのような影響を与えるかを理解します。 FDA のコンプライアンス要件、ALCOA+ の原則、監査失敗を防ぐ実証済みの戦略について学びます。

分析テストで OOS (規格外) の結果を調査する方法を学びます。 製薬コンプライアンスのための根本原因分析、FDA ガイドライン、CAPA 戦略を理解します。


















AI とノーコード プラットフォームが、よりスマートな品質管理、予知保全、検査対応の文書化によって製薬製造をどのように変革しているかを探ります。

Discover the hidden operational debt pharma companies face by delaying low-code and AI adoption—and how AmpleLogic helps reduce it with GxP-compliant workflows.

AmpleLogic を使用して年次製品品質レビューを自動化します。 準備時間を 70 ~ 80% 削減し、GMP 準拠を確保し、規制対応の PQR レポートをより迅速に生成します。

AmpleLogic RIMS を使用して申請、更新、CMC 更新、グローバル申請を正確に管理するために、製薬規制チームが RIMS ソリューションを必要とする理由を学びましょう。

AI を活用したソリューションでコンプライアンスを確保し、手動エラーを排除し、データの整合性を向上させるために、製薬工場に eLogbook が必要な理由を説明します。

AmpleLogic AI は、医薬品逸脱の処理時間を短縮し、根本原因分析を自動化し、コンプライアンスを向上させ、調査を迅速化します。

製薬業界での洗浄検証に関する AmpleLogic ガイドは、GMP 準拠を保証し、汚染を防止し、製造効率を向上させます。


エーザイ製薬、AmpleLogic 従業員トレーニング管理ソフトウェアの導入に成功

Hetero Labs Limited が AmpleLogic キャリブレーションおよび予防メンテナンス ソフトウェアを稼働開始













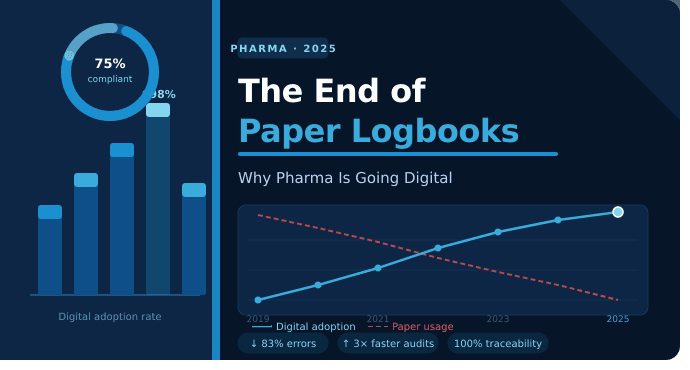
製薬メーカーであるあなたは、業界を統治する厳しい規制要件をよくご存じだと思います。 コンプライアンスの重要な側面の 1 つは、正確かつ最新の記録と文書を維持することです。 しかし、あなたの記録管理システムにおけるログブックの役割を考慮したことがありますか? そうでない場合は、始めてみるとよいでしょう。

ライフ サイエンス業界にデジタル テクノロジーを組み込むことは、最近のビジネス上の必須事項となっています。 日々の規制の更新、文書の増加、より迅速な監査の必要性により、製薬会社は着実に手動プロセスからの移行を進めています。 そのような変化の 1 つは、コンプライアンス、効率、データの整合性を確保するために不可欠になりつつあるツールである文書管理システム (DMS) への移行です。 デジタルイノベーションはあらゆる分野に影響を及ぼしていますが、製薬業務と品質保証におけるデジタルイノベーションの役割は特に変革をもたらします。 業界を変革する数多くのツールの中でも、DMS は重要なソリューションとして際立っており、ドキュメントのワークフローを自動化し、紙への依存を減らし、重要なドキュメントの集中管理を提供します。

電子学習管理システム ソフトウェアを通じた e ラーニングの人気が高まり、さまざまな業界、特に製薬分野で大きな注目を集めています。 製薬産業は、ヨーロッパやその他の経済において最も重要なセクターの 1 つとして、近年急速な成長を遂げています。 パンデミック後、多くの国で医療とワクチン接種が最優先事項になりました。 しかし、その緊急性は市場にある他の必須医薬品にも及びました。 このペースの速い環境において、医薬品 LMS は、新規人材のトレーニングや認定資格の提供において、従業員が業界の増大する需要に対応できる準備が整っていることを確認するための重要な機能となっています。 製薬業界が拡大し続けるにつれて、熟練した労働力の必要性が着実に高まっています。 最新のMarketsandMarketsレポートによると、

ファーマ 4.0 という用語は、もはやカンファレンスで議論される概念ではありません。 それはインドの現場で実用化されています。 製薬会社は製剤ラボや製造部門全体で、自動化、データの可視化、よりスマートなインフラストラクチャを導入して、時代遅れのシステムを着実にアップグレードしています。 この移行により、生産の遅延、データ整合性の欠如、手動への依存、規制の監視の強化など、地元の業界の問題が解決されます。 インドの製薬業界がデジタル化と先進技術を受け入れるにつれて、いくつかの必要な変革が進行中です。






















今日の高度に規制されたライフサイエンス環境では、組織は俊敏性と厳格なコンプライアンス要件のバランスを取る必要があります。 ローコード QMS および MES ソリューションは、大規模なコーディングを行わずに迅速なアプリケーション開発を可能にすることで、製薬会社やバイオテクノロジー企業が品質、製造、規制のワークフローを管理する方法を変革しています。 AmpleLogic のローコード プラットフォームは、品質管理、製造実行、実験室システム、規制プロセスを単一のデータ駆動型フレームワークに統合する統合エコシステムを提供します。 これによりサイロが排除され、リアルタイムの可視性が向上し、業務全体にわたる迅速な意思決定が可能になります。 FDA 21 CFR Part 11、GxP、Annex 11 などのコンプライアンス フレームワークが組み込まれているため、組織は監査の準備を整えながら、展開のタイムラインを数か月から数週間に短縮できます。 このプラットフォームのドラッグ アンド ドロップ構成、自動化されたワークフロー、AI を活用した洞察により、手動作業が軽減され、データの整合性が向上し、継続的な改善が促進されます。 ローコードの QMS および MES ソリューションを採用することで、企業は運用の機敏性を高め、コストを削減し、一貫した規制遵守を維持することができ、ライフ サイエンス業界における現代のデジタル変革にとって重要な戦略となっています。

製薬業界はデジタル革命の入り口に立っています。インダストリー4.0テクノロジーは医薬品の製造、試験、世界的な流通を変革し、より効率的で柔軟かつコンプライアントなスマートファクトリーを創出しています。

Laboratory Information Management System(LIMS)は、現代の製薬試験室においてワークフローを効率化し、コンプライアンスを強化し、効率性を向上させるための不可欠なツールです。

製薬クリーニングバリデーションにおける規制コンプライアンスは非交渉的です。適切なプロトコルの理解と実装は、製品の安全性と製造の整合性を確保します。

製薬製造においてクリーニングバリデーションのベストプラクティスを実装することは、製品品質の維持、交差汚染の防止、GMPコンプライアンスの確保に不可欠です。

適切なLIMSソフトウェアの選定は、製薬試験室にとって重要な決断です。適切なシステムは試験室業務を変革し、コンプライアンスを強化し、結果達成時間を短縮します。

AIと機械学習の製造実行システムへの統合は、これまで不可能だった予測分析とリアルタイム最適化機能で製薬製造を変革しています。

長期計画は、複雑な規制環境、変化する市場ダイナミクス、技術変革をナビゲートする製薬会社にとっての戦略的必須事項です。

aPaaS(Application Platform as a Service)とCOTS(市販品)ソリューションおよびローコード開発の融合が、製薬会社のアプリケーション構築・展開方法を再形成しています。

製薬・バイオテクノロジー業界では競争が非常に激しく、品質基準への要求は厳しさを増しています。製造に関わる工程の継続的な監視と調査が求められます。

製薬業界やその他の様々な分野において、環境条件の継続的な監視は規制基準の遵守と製品品質の確保のために最重要です。

製薬業界は、規制要件とデータインテグリティおよびコンプライアンス強化の必要性に駆られ、ペーパーレスバッチ製造記録(BMR)へと移行しつつあります。

製薬業界におけるクリーニングバリデーションは、製品品質を維持し交差汚染を防止するために規制当局が定める必須のプロセスです。

今日の急速に変化する高度に規制された業界において、製品レビューソフトウェアは不可欠なツールとなっています。基本的なトラッキングシステムから高度で統合されたプラットフォームへと進化しています。

業務上の卓越性と品質保証の追求において、確立された規範からの逸脱は予想されるものです。成功する組織を真に差別化するのは、これらの逸脱をどのように処理するかです。
%20Software.webp&w=3840&q=80)
今日の競争激しいビジネス環境において、高い製品品質を維持し継続的に改善することは成功に不可欠です。APRソフトウェアは強力なツールとして台頭しています。

ダイナミックで高度に規制された製薬製造の世界では、高度なテクノロジーの統合が主要な業務プロセスの向上と標準的な製品品質の確保に不可欠です。

年次製品品質照査(APQR)の重要な側面を探求するこの調査では、この重要な品質管理プロセスを定義する「何を」「なぜ」「どこで」という基本的な問いを掘り下げます。

製薬業界において、バッチ製造記録(BMR)のバリデーションは製品品質、安全性、規制コンプライアンスを確保するために不可欠です。従来、このバリデーションは手作業によるプロセスでした。

急速に進化する製薬業界において、資産の効率的な管理は最重要です。研究室から製造施設、流通ネットワークまで、製薬会社は膨大な設備とインフラに依存しています。

製薬業界において、データの整合性は最重要です。ALCOA原則—帰属可能、判読可能、同時代的、原本、正確—は製造におけるデータインテグリティを確保するために確立されました。

学習管理システム(LMS)は製薬業界においてますます重要となっており、トレーニング、コンプライアンス、専門能力開発を管理するための体系的で効率的な方法を提供しています。
-in-Equipment.webp&w=3840&q=80)
効率性と精度は、ライフサイエンスおよび製薬業界の本質的な側面です。従来の手作業プロセスからの脱却による業務プロセスの卓越性実現を支援します。

年次製品品質照査(APQR)は、製薬会社が製品の品質基準を評価し、規制コンプライアンスを確保し、継続的改善を推進するための重要なプロセスです。人工知能はこの照査の実施方法を革新しています。

変更管理は、製薬、医療機器、食品・飲料などの規制産業における重要なプロセスです。製品、工程、システムへの変更を管理・文書化することを含みます。

設備保全は、製薬製造・生産部門の効率性と生産性を確保するための重要な側面です。設備保全ソフトウェアはこの分野を革新しています。

精度、コンプライアンス、効率性が重要資産である複雑な製薬製造の世界では、企業は資産管理の複雑さを乗り越える革新的なソリューションを求めています。

業務卓越性の追求が戦略的必須事項であり持続的成功の礎である、複雑で常に進化する現代産業において、APQR分析は中心的な役割を果たしています。

製薬業界において、製品品質、患者安全、規制コンプライアンスを確保するためにデータインテグリティの維持は不可欠です。電子ログブックはALCOA+準拠の維持に必要不可欠なツールです。
-Software-in-Deviation-Handling.webp&w=3840&q=80)
効率性、正確性、コンプライアンスは製薬製造業界の本質的な要素です。標準操作手順からのいかなる逸脱も製品品質を損なう可能性があります。

根本原因分析(RCA)は、品質プロセスにおける問題や不適合の「根本原因」(発生の実際の原因)を特定するために使用される手法です。RCAは原因の是正・排除と再発防止に役立ちます。

製薬トレーニング管理システム(TMS)は、GMPコンプライアンストレーニング、コンピテンシー追跡、規制監査対応を効率化します。最新のLMSがトレーニングコストを最大50%削減し、従業員の資格認定を加速させ、グローバルオペレーション全体で21 CFR Part 11準拠を確保する方法をご紹介します。

電子文書管理システム(eDMS)は、製薬・ライフサイエンス企業の文書管理プロセスを根本的に変革しました。しかし、FDA 21 CFR Part 11への準拠や明確なコンプライアンス上の利点にもかかわらず、多くの組織がこのデジタル変革を完全に受け入れることをためらっています。

製薬会社が受けてきた多数のデータインテグリティ警告により、問題発生を防ぐための追跡メカニズムの構築・強化に注力が集まっています。解決策の一つはデジタル化ですが、現在の課題はデジタルトランスフォーメーションが期待より遅く進んでいることです。
製薬製造、コンプライアンス、デジタルトランスフォーメーションの最新トレンドをお届けします。
必要な場所に、私たちがいます。グローバルにつながる、ローカルに届ける。
Melange Tower, 2nd Floor, Wing-C, Patrika Nagar, HITEC City, Madhapur, Hyderabad - 500081, Telangana, India
5255 Yonge Street Suite 201, North York, ON, M2N 6P4
Block 1, Blanchardstown Corporate Park Ballycoolen Road, Dublin, D15 AKK1
1 Scotts Road, #24-10 Shaw Centre, Singapore, Singapore, 228208
最新の製品アップデート、コンプライアンスニュース、業界インサイトをお届けします。