

General5 min read
From Drug Discovery to Commercial Operations: How AmpleLogic Powers the Entire Pharmaceutical Value Chain
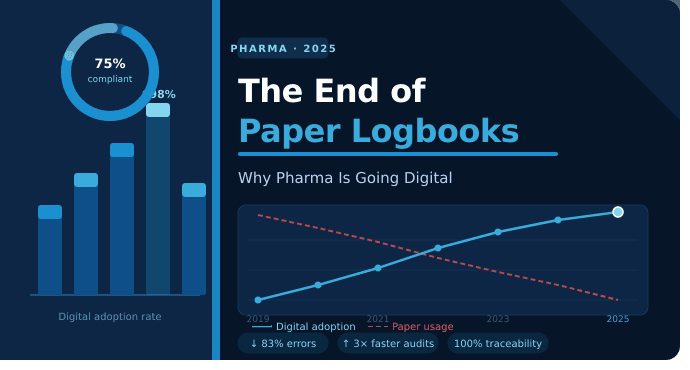
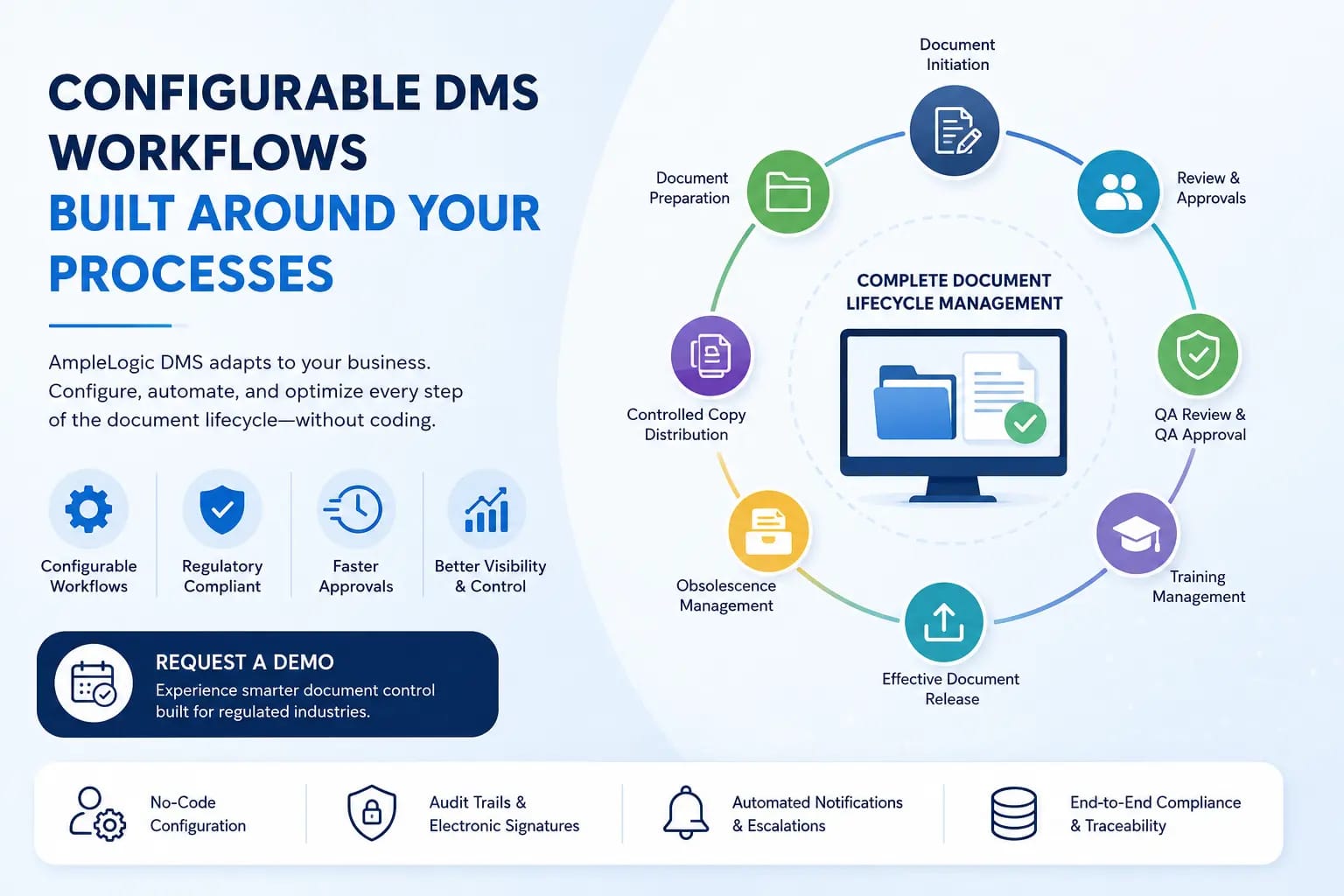
Most pharma companies run five to ten disconnected systems across research, quality, manufacturing, and regulatory functions. Here's how a single connected platform changes what's possible across the entire product lifecycle.
2026-07-30Read more













































































































































































































































































































%20Software.webp&w=3840&q=80)






-in-Equipment.webp&w=3840&q=80)






-Software-in-Deviation-Handling.webp&w=3840&q=80)



